- The paper demonstrates that binding sfTA reduces finite size error, lowering mean absolute error in binding energies from 20 meV/atom to as low as 4 meV/atom.
- It introduces paired and binding sfTA variants that ensure matched twist angles between monolayer and bilayer calculations, enabling effective error cancellation.
- The method significantly cuts computational cost by approximating twist-averaged CCSD results through a single high-level calculation, broadening applications in 2D materials.
Structure Factor Twist Averaging Adaptations for Bilayer Electronic Structure
Introduction
Accurate modeling of electron correlation in low-dimensional materials is essential for quantifying interlayer binding energies and exploring novel physics in systems such as bilayer graphene. This paper addresses the challenge posed by finite size errors (FSEs) in periodic electronic structure calculations, particularly for demanding methods like CCSD, which are prohibitively expensive with conventional twist-averaging (TA). The recently developed structure factor twist averaging (sfTA) method offers a substantial reduction in computational cost for bulk materials, but its applicability to low-dimensional systems and interaction energies has not been systematically explored. The authors introduce and evaluate two sfTA variants—paired sfTA and binding sfTA—tailored for bilayer systems, comparing their binding correlation energies to TA benchmarks across an extensive variety of 2D materials.
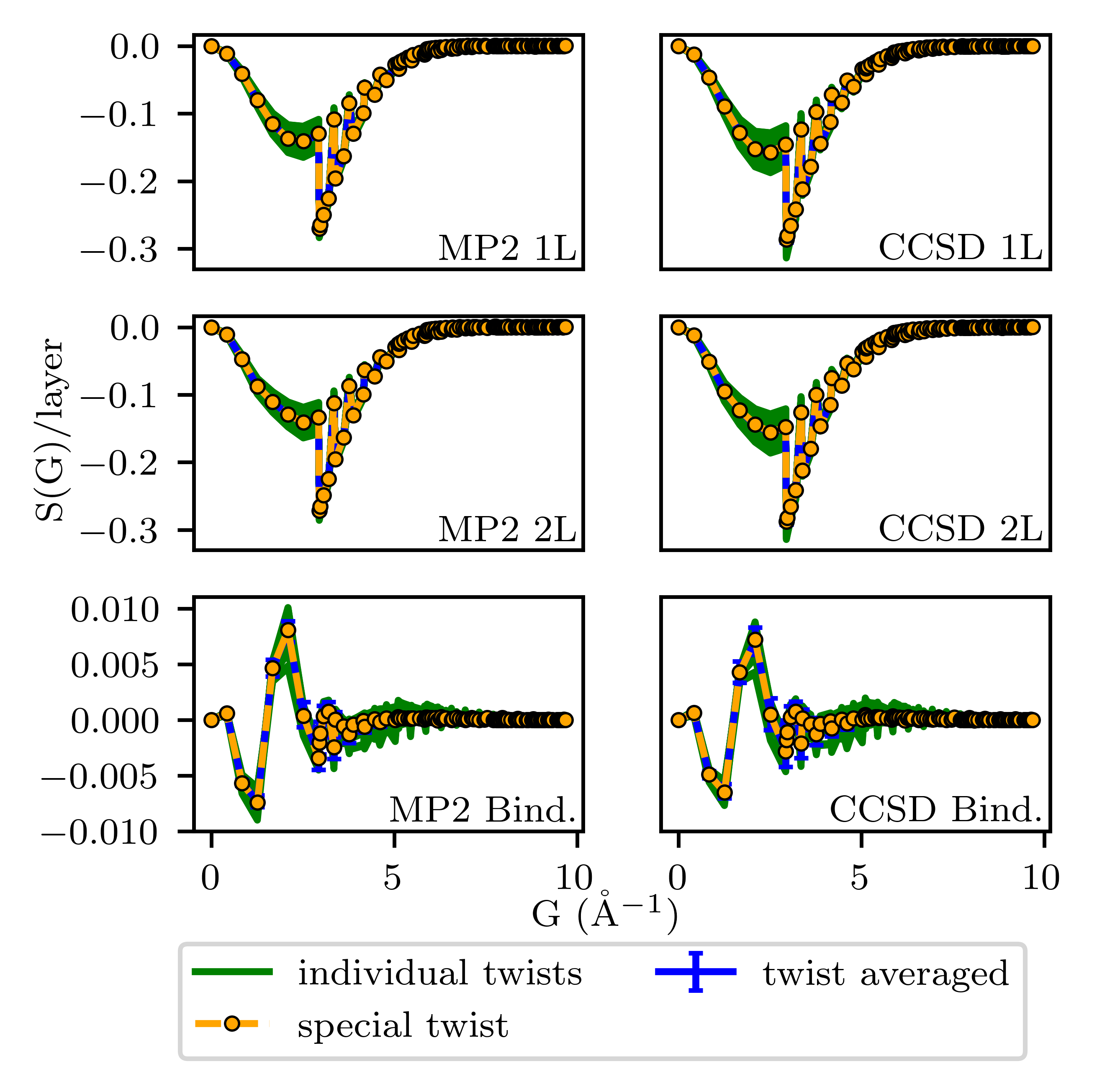
The electronic correlation is computed using MP2 as the low-level method and CCSD as the high-level reference. The correlation energy is formulated via Tijabvijab transitions and the transition structure factor S(G), following the Liao-Grüneis framework. sfTA selects a "special" twist angle by minimizing the residual between the transition structure factor at each random twist and the twist-averaged structure factor. This enables a single high-level calculation to approximate TA.
sfTA Variants for Bilayer Interactions
Numerical Results and Validation
Tests are conducted on two classes of systems: a small set (e.g., graphene, BN, SiC, Si, GaN) and a larger, more challenging set (including metallic and semiconducting dichalcogenides, Bi, Sn, Pb). For all three methods—original sfTA, paired sfTA, and binding sfTA—monolayer, bilayer, and binding correlation energies are evaluated against TA references.
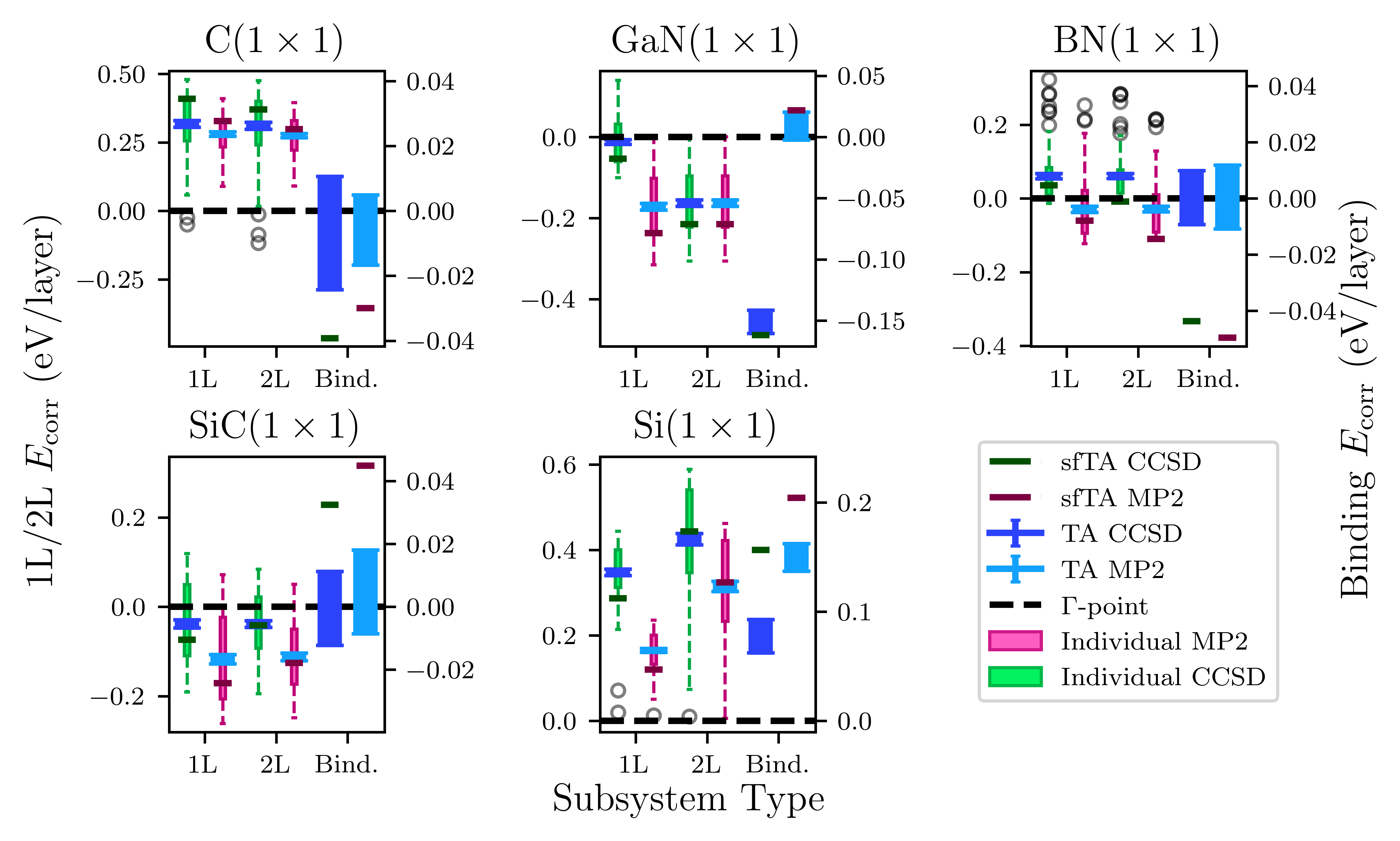
Figure 2: Boxplots of 1L, 2L, and binding energies for original sfTA versus TA. sfTA deviates notably from TA for binding energies.
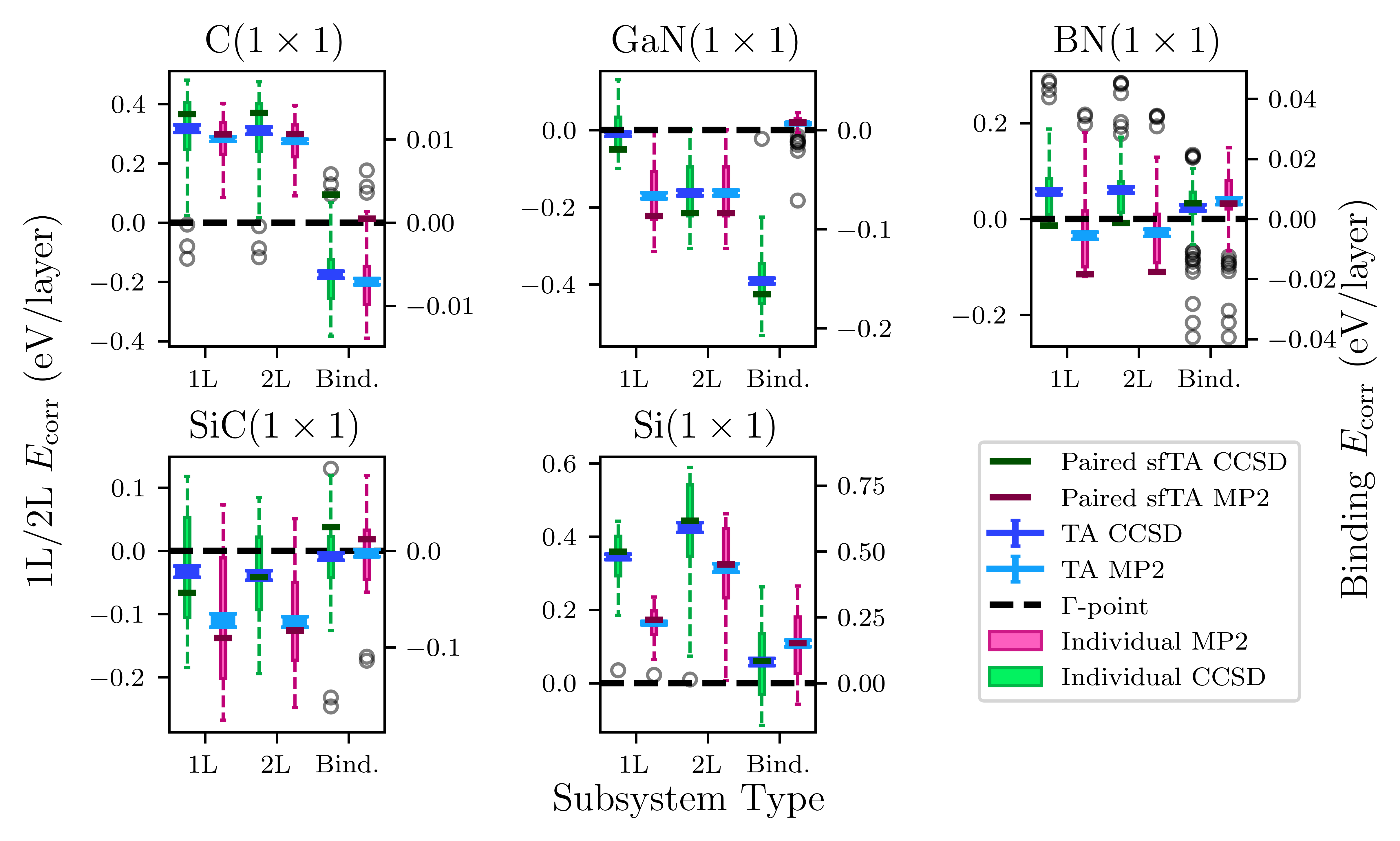
Figure 3: Paired sfTA exhibits reduced error in binding energies compared to original sfTA; 1L and 2L errors remain largely unchanged.
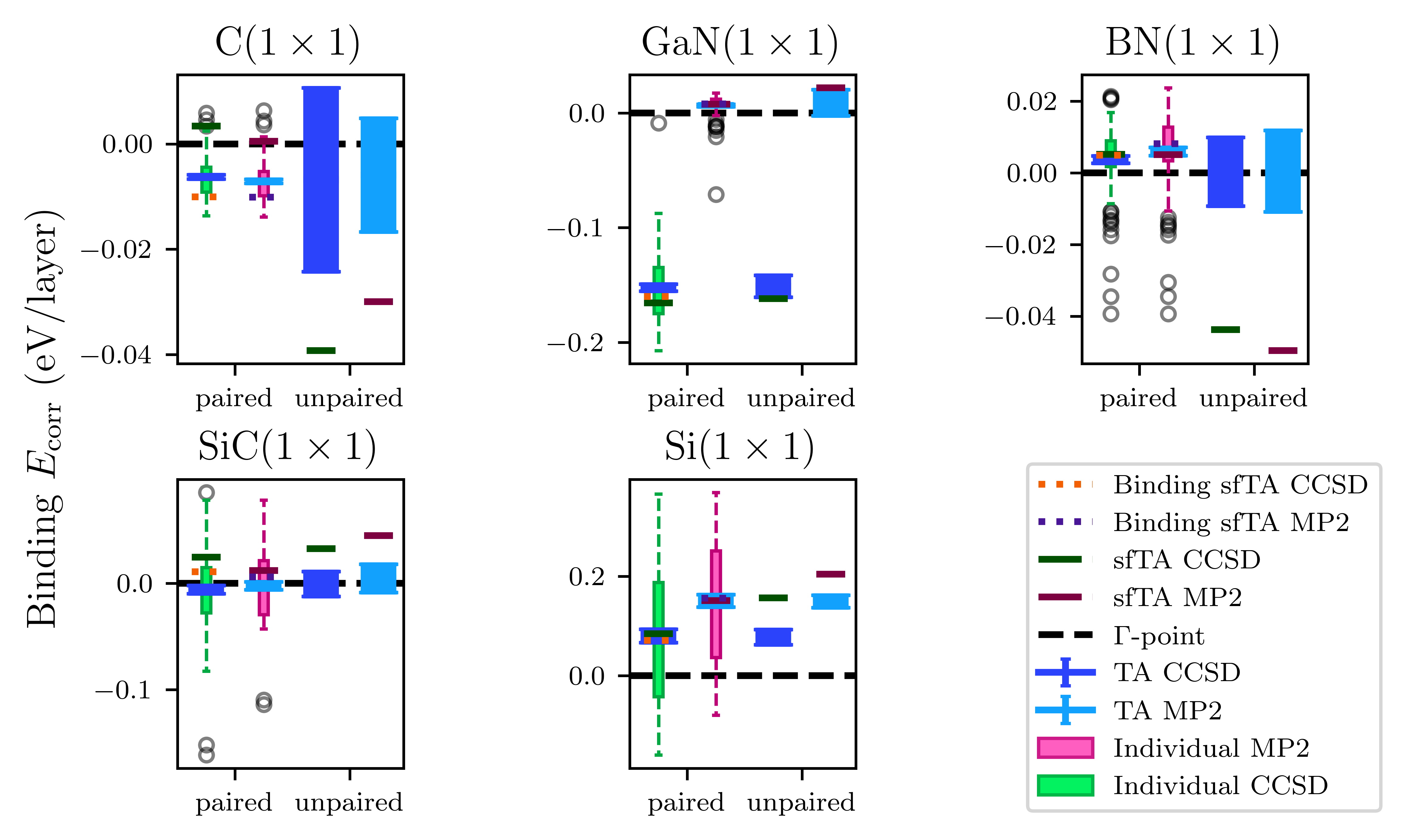
Figure 4: Comparison of binding energies across sfTA variants; binding sfTA yields the closest results to TA.
Numerically, the mean absolute error (MAE) in the small test set for binding energies drops from 20(3) meV/atom (original sfTA) to 6(1) (paired sfTA), and further to 4(1) meV/atom (binding sfTA).
In the challenging set, binding sfTA achieves an MAE of 10(1) meV/atom, outperforming paired sfTA (16(1) meV/atom). The improvement is consistent across materials with disparate electronic structure.
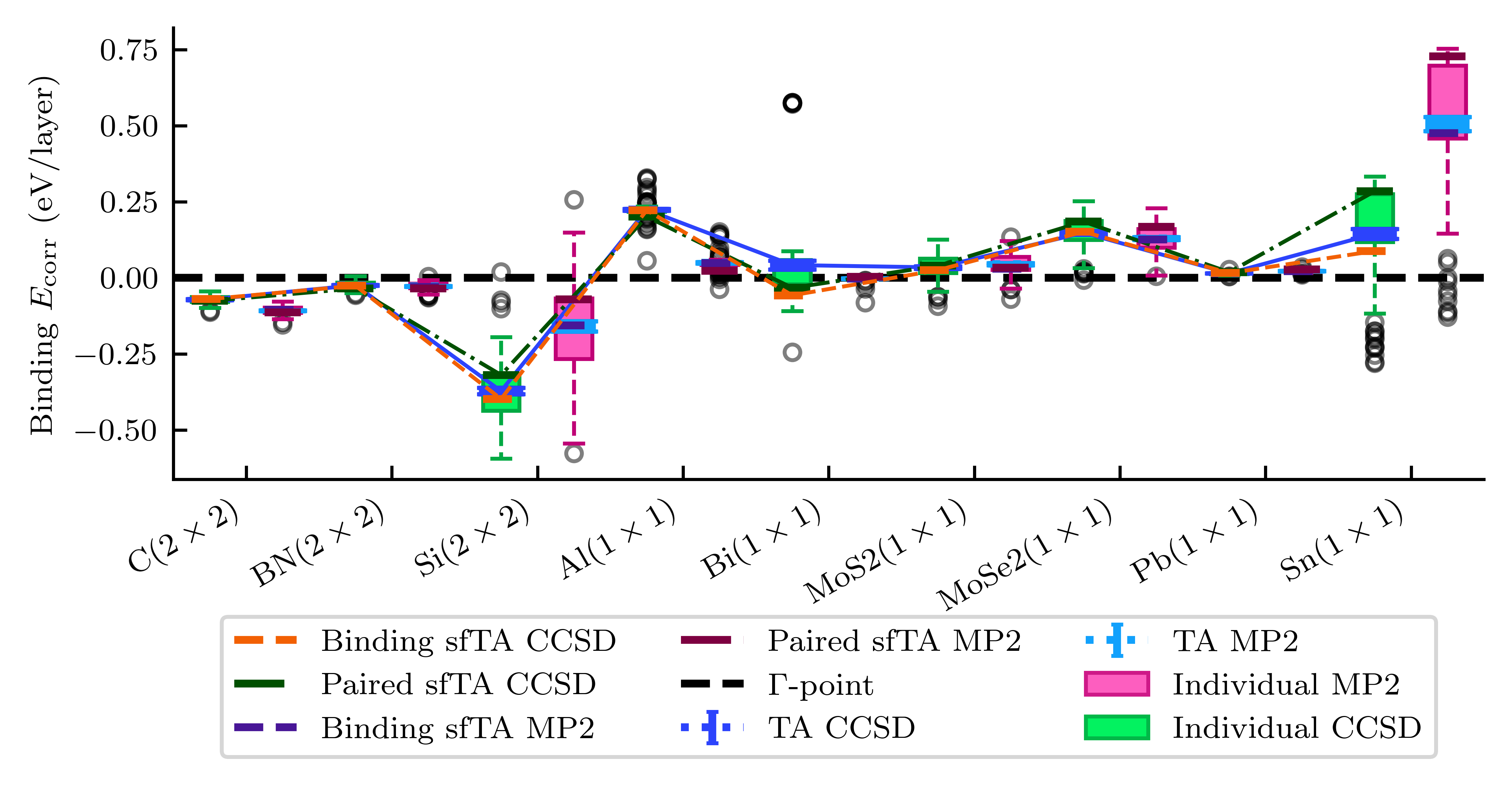
Figure 5: Binding energies for challenge systems; binding sfTA consistently tracks the TA reference more closely than paired sfTA.
Error Landscape and Twist-Angle Selection
Contour plots for 1×1 graphene illustrate energy landscapes as functions of 2D twist angle, revealing pronounced error cancellation in binding calculations when twist angles are held constant between 1L and 2L systems.
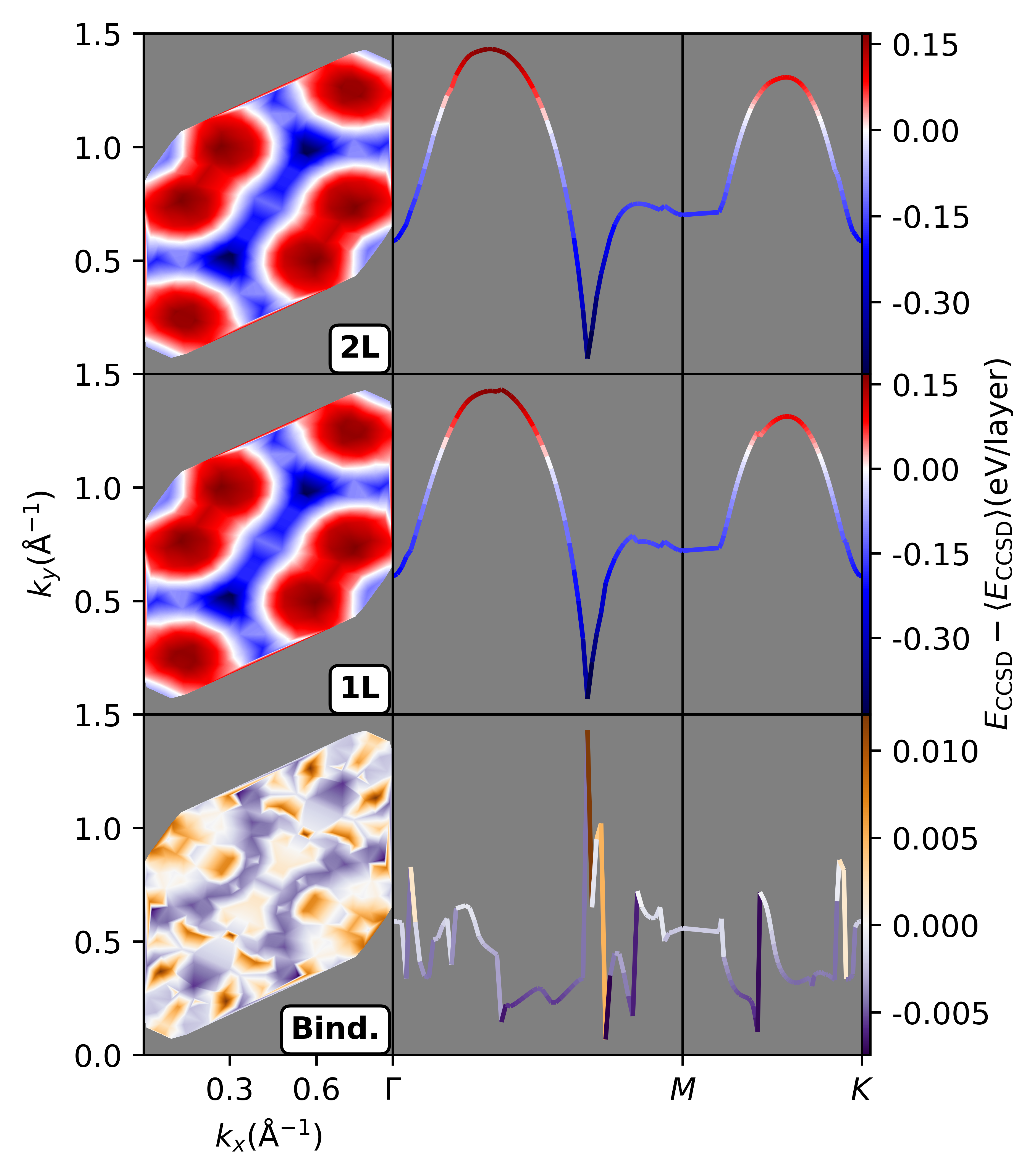
Figure 6: Contour plots of CCSD correlation energy versus twist angle for 1L, 2L, and binding configurations; binding energy is flatter and consistently near TA due to cancellation of twist-induced errors.
The error cancellation effect ensures that even suboptimal twist angles for 1L or 2L produce binding energies near TA, provided that the twist angle is held fixed. High-symmetry points (e.g., Γ) are poor choices for total energy but become highly accurate for binding, as the dominant FSEs cancel. The maximal deviation from TA is reduced drastically in the binding landscape compared to isolated layers.
Theoretical and Practical Implications
The results assert that for bilayer interaction energies, constraining the twist angle between monolayer and bilayer calculations is critical. Binding sfTA, by explicitly incorporating the binding structure factor, systematically improves TA agreement. As a result, expensive CCSD TA calculations can be bypassed in favor of a single high-level computation at an optimally chosen twist, effecting a near 100-fold reduction in computational resources.
The observed error cancellation mechanism has broader implications. It is plausible that similar adaptations could extend to other interaction-driven systems in reduced dimensionality, such as adsorption on 2D surfaces or further van der Waals heterostructures. The structure factor's utility in controlling FSEs paves the way for accurate yet affordable ab initio studies of correlated low-dimensional electronic phenomena.
Open questions remain concerning alternate twist-selection criteria based on connectivity or spectral properties, which may yet further refine energy accuracy or adaptability.
Conclusion
The paper demonstrates that paired sfTA and binding sfTA variants improve the accuracy of interlayer binding energies in bilayer materials relative to original sfTA, with binding sfTA delivering the best performance. The theoretical foundation for this improvement is shown to be error cancellation across matched twist angles, as visualized by twist-angle energy contours. The binding sfTA method generalizes the utility of structure factor-based twist averaging to low-dimensional, interaction-dominated materials, enabling high-level correlation calculations with minimal computational overhead. This development is anticipated to accelerate quantitative investigations of 2D material assemblies and interaction energies in broader contexts (2604.23405).