- The paper introduces ProDock as a reproducible workflow that integrates preparation, multi-target docking, postprocessing, and persistent storage for improved experimental traceability.
- It implements a modular four-stage pipeline—preprocessing, provenance-aware execution, postprocessing, and SQLite-based data management—to deliver auditable and standardized results.
- The EGFR screening case study demonstrates ProDock’s automated handling of configurations and consensus docking, establishing a scalable framework for large-scale drug discovery campaigns.
ProDock: A Reproducible Workflow for Structure-Based Docking and Data Management
Introduction
The paper "ProDock: From multi-target consensus docking into database-backed storage" (2604.21828) presents ProDock, an open-source Python toolkit that addresses the reproducibility and traceability gaps inherent in application-level structure-based docking workflows. Unlike existing solutions that focus on scoring algorithms or search heuristics, ProDock centralizes the ancillary yet critical elements of docking—preparation, provenance, and data management—into a unified architecture. This addresses longstanding concerns regarding workflow fragmentation, result provenance, and analytical scalability in large-scale structure-based drug discovery campaigns.
Workflow Design and Architecture
ProDock operationalizes the protein–ligand docking process through a reproducible, project-local, four-stage workflow: (1) preprocessing of receptors and ligands, (2) structured docking execution, (3) analytical postprocessing, and (4) persistent storage with SQLite-backed querying. This modular architecture is realized in Python and is designed for direct extensibility by computational chemists and informatics researchers.
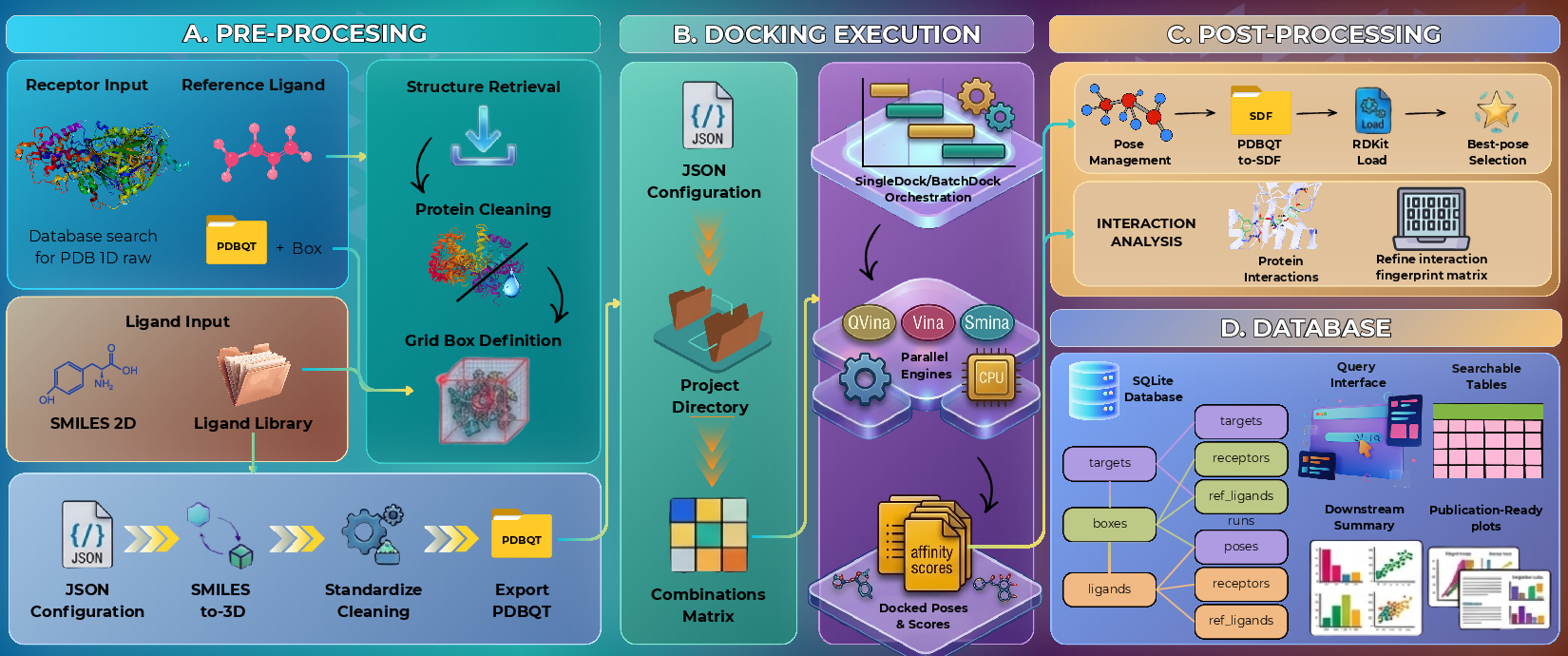
Figure 1: The ProDock workflow consists of preprocessing, docking execution, postprocessing, and SQLite-based data storage, forming a coherent and reproducible pipeline for structure-based docking.
Stage 1: Preprocessing
ProDock's preprocessing modules explicitly manage receptor and ligand setup, using standard resources (e.g., RCSB PDB entries, SMILES libraries) and robust toolkits such as RDKit, Open Babel, PDBFixer, and Meeko. By formalizing this stage, the toolkit ensures structural standardization and data quality; prepared inputs become persistent, versioned artifacts rather than ephemeral intermediates, mitigating silent errors from malformed inputs or inconsistent box definitions. Automatic grid box determination via integrated algorithms further reduces manual specification errors and improves experimental provenance.
Stage 2: Provenance-Aware Docking Execution
Docking operations are organized as explicit, multi-receptor, multi-ligand, and multi-backend "campaigns". ProDock decouples engine-specific execution logic, enabling seamless interchange between supported backends (e.g., Smina, Vina, QuickVina, GNINA) and facilitating many-to-many experimental mappings critical for consensus and benchmarking studies. All campaign parameters—ranging from random seeds to engine-specific options—are serialized in machine-readable JSON, providing an exhaustive, auditable provenance record.
Stage 3: Postprocessing and Interaction Fingerprinting
The postprocessing layer parses raw output from heterogeneous docking engines, extracts pose and score information in a normalized format, and generates both compact and expanded summaries. This includes per-pose score tabulation and residue-level interaction fingerprinting using the integrated ProLIF toolkit. The coupling of geometrical analysis with cheminformatic profiling enables downstream structure-activity relationship (SAR) exploration, binding mode validation, and automated comparison across ligands, engines, and targets.
Stage 4: Database-Backed Storage and Querying
A key innovation is the persistent, normalized SQLite schema underpinning ProDock's storage layer. The schema decouples project metadata (receptors, ligands, engines) from factual records (poses, scores, residue-level interactions), ensuring analytical flexibility, high data integrity, and the ability to perform post hoc analyses without reparsing or manual curation. The database-centric approach enables efficient querying, complex filtering, and statistical analysis, accelerating cross-campaign comparisons and large-scale method benchmarking.
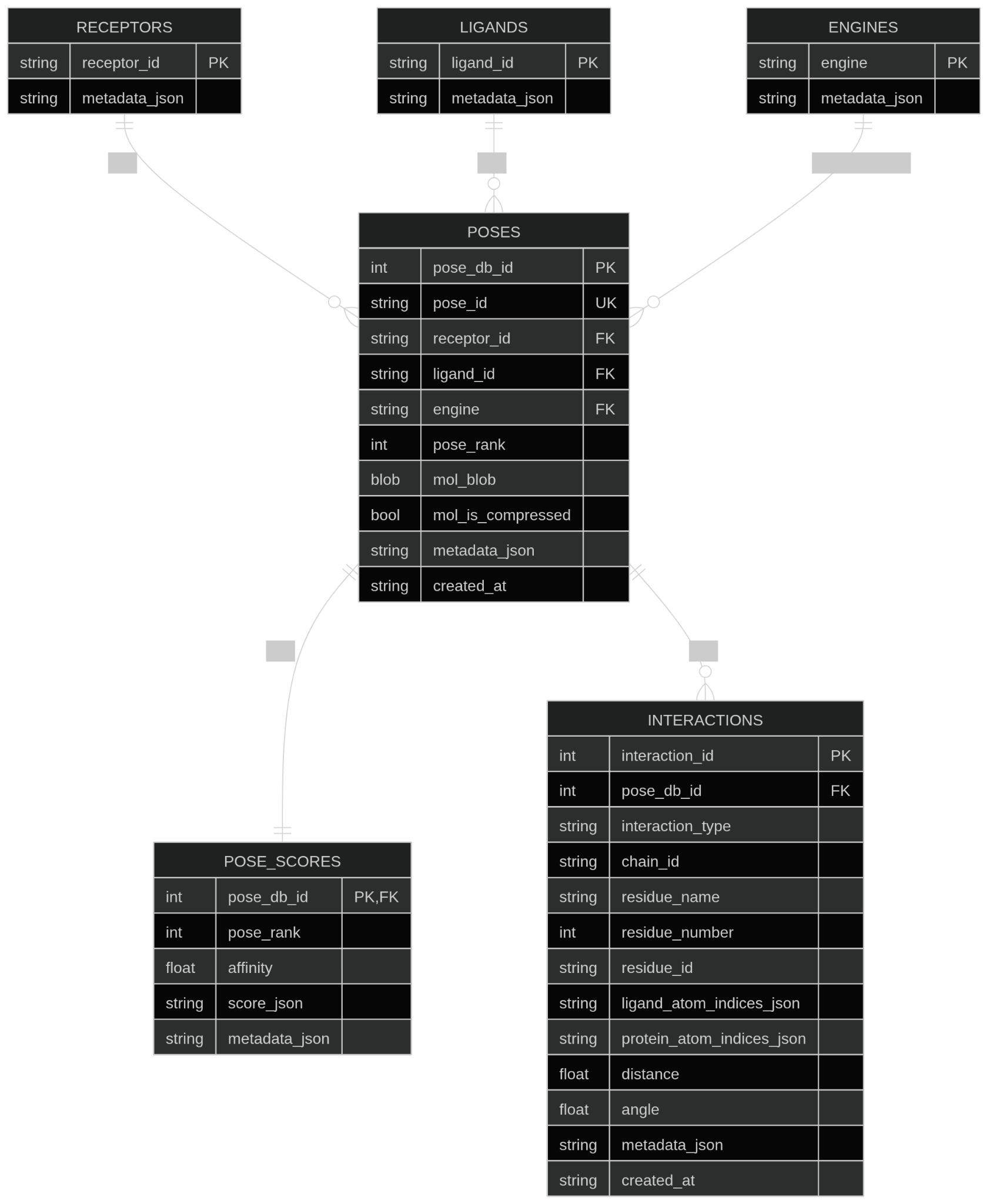
Figure 3: ProDock’s SQLite schema, which manages dimension tables for receptors, ligands, and engines, and captures docking geometries, scores, and residue-level interactions in normalized factual tables.
Reproducible Project Layout and Use
Adopting a split-configuration model, ProDock enforces a consistent directory structure and explicit separation between configuration, receptor, and ligand specifications. Campaign metadata and all workflow artifacts—including prepared molecules, logs, and results—are co-located within a project-specific directory, optimizing both reproducibility and collaborative handover. The workflow is accessible programmatically via the Python API and from a command-line interface, facilitating both interactive and batch processing modes.
The project data model, including campaign JSON and SQLite database, provides immediate transparency for both completed and archival runs. This architecture ensures unambiguous traceability of how every pose and score was generated, directly addressing reproducibility deficits in conventional docking pipelines.
Applied Case Study and Utility
As a practical demonstration, the authors report an EGFR screening campaign involving five crystal structures and a curated ligand library, showcasing the autonomous system-preparation, batch campaign execution, and data consolidation features of ProDock. Without manual intervention in box definition or intermediate scripting, the framework handled retrieval, cleaning, box inference, batch docking across four engines, and full interaction fingerprint extraction. The entire experiment was archived in a queryable, self-contained directory—supporting immediate post hoc analysis and reuse.
Implications and Future Developments
ProDock’s core contribution is its explicit, end-to-end formalization of docking workflows, offering structural bioinformatics researchers a reproducible and fully auditable path from molecular inputs to analytical outputs. Its many-to-many data model supports varied and realistic experimental designs, such as consensus scoring, multi-target campaigns, and retrospective benchmarking.
This work establishes a new standard for transparency, provenance, and data management in computational docking. It could facilitate more rigorous method development (by permitting robust comparative studies), streamline collaborative benchmark datasets, and provide a modern, extensible foundation for automated and ML-driven structural discovery pipelines.
Potential future extensions include integration with cloud or distributed compute infrastructures, support for alternative backend engines (e.g., deep learning–based scorers), direct visualization modules, or tighter feedback loops with generative or optimization frameworks in drug design. As the scale and complexity of screening studies increase, robust workflow-level solutions like ProDock will become increasingly essential.
Conclusion
ProDock addresses a critical infrastructure gap in structure-based docking by unifying preparation, docking, postprocessing, and result storage within a coherent, extensible, and reproducible workflow (2604.21828). Its architecture enables many-to-many and consensus-based designs, robust provenance tracking, and analytical flexibility via normalized database storage. Rather than introducing new scoring functions, it professionalizes the management of computational docking studies, enabling greater methodological rigor and facilitating transparent, collaborative research in molecular discovery.