- The paper presents a high-throughput DFT analysis of over 2,300 diastereomeric transition states, revealing significant barrier splitting that impacts autooxidation kinetics.
- The study details a robust workflow that propagates stereochemical resolution from hydrocarbon precursors to validated cyclic transition states using DFT optimization and IRC validation.
- Results underscore that neglecting stereochemical distinctions in automated mechanism generation can lead to underestimations of reaction rates in combustion models.
Chemical Space Analysis of Diastereomeric Barriers in Alkylperoxy-to-Hydroperoxyalkyl Isomerization
Introduction
The investigation "A Chemical Space Perspective on Diastereomeric Barriers in Alkylperoxy-to-Hydroperoxyalkyl Isomerization" presents a high-throughput computational analysis of the influence of stereochemistry on the reactivity of hydrocarbon intermediates in low-temperature autooxidation. The principal contribution is the Stereochemically Expanded Autooxidation Reaction Space (SEARS), a dataset comprising DFT-level characterizations of over 5,000 species, including 2,324 validated cyclic diastereomeric transition states for the pivotal ROO∙→∙QOOH isomerization. The work emphasizes that explicit stereochemical resolution is essential for mechanistically faithful kinetic modeling in automated combustion mechanism generation frameworks, as constitutionally collapsed molecular representations systematically omit kinetically relevant channels.
Workflow and Data Generation
A robust workflow was executed to propagate stereochemical information throughout the autooxidation sequence, starting from 498 nonaromatic C1–C7 hydrocarbons. Following rigorous filtering and enumeration of stereoisomeric and diastereomeric parent structures, stereochemically distinct alkyl, peroxy, and hydroperoxyalkyl radicals were generated in SMILES format with full conformational and diastereotopic resolution.
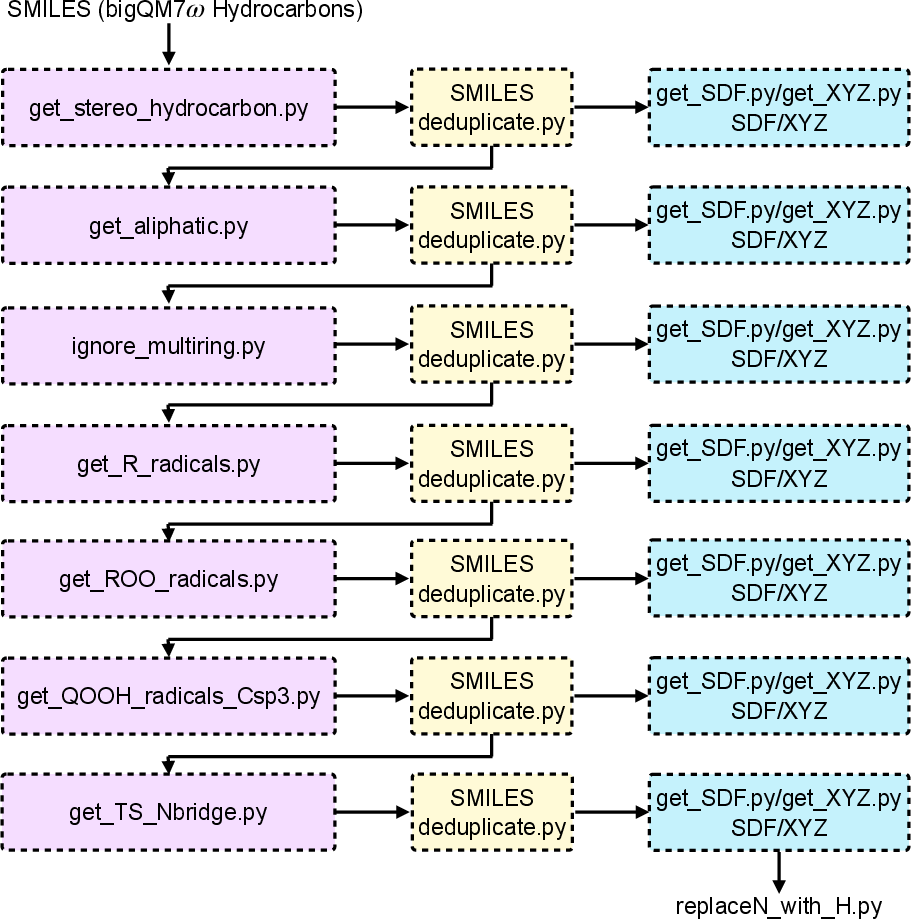
Figure 1: Workflow for the stereochemical enumeration of starting hydrocarbons through radical and peroxy intermediates to diastereomeric transition-state pairs for ROO∙→∙QOOH isomerization.
Successive automated protocols constructed 3D initial geometries for all species, leveraging temporary N-bridging for cyclic hydrogen-abstraction transition states to facilitate realistic conformational guessing, subsequently replaced with H for DFT calculations. The sequence yielded 5,590 candidate transition-state connectivities, with 3,808 viable 3D geometries selected for quantum chemical calculations.
Subsequent DFT optimization at the ωB97M-D4/def2-SV(P) level furnished stationary points, which were subjected to intrinsic reaction coordinate (IRC) validation to confirm proper connectivity of transition states and product/reactant wells.
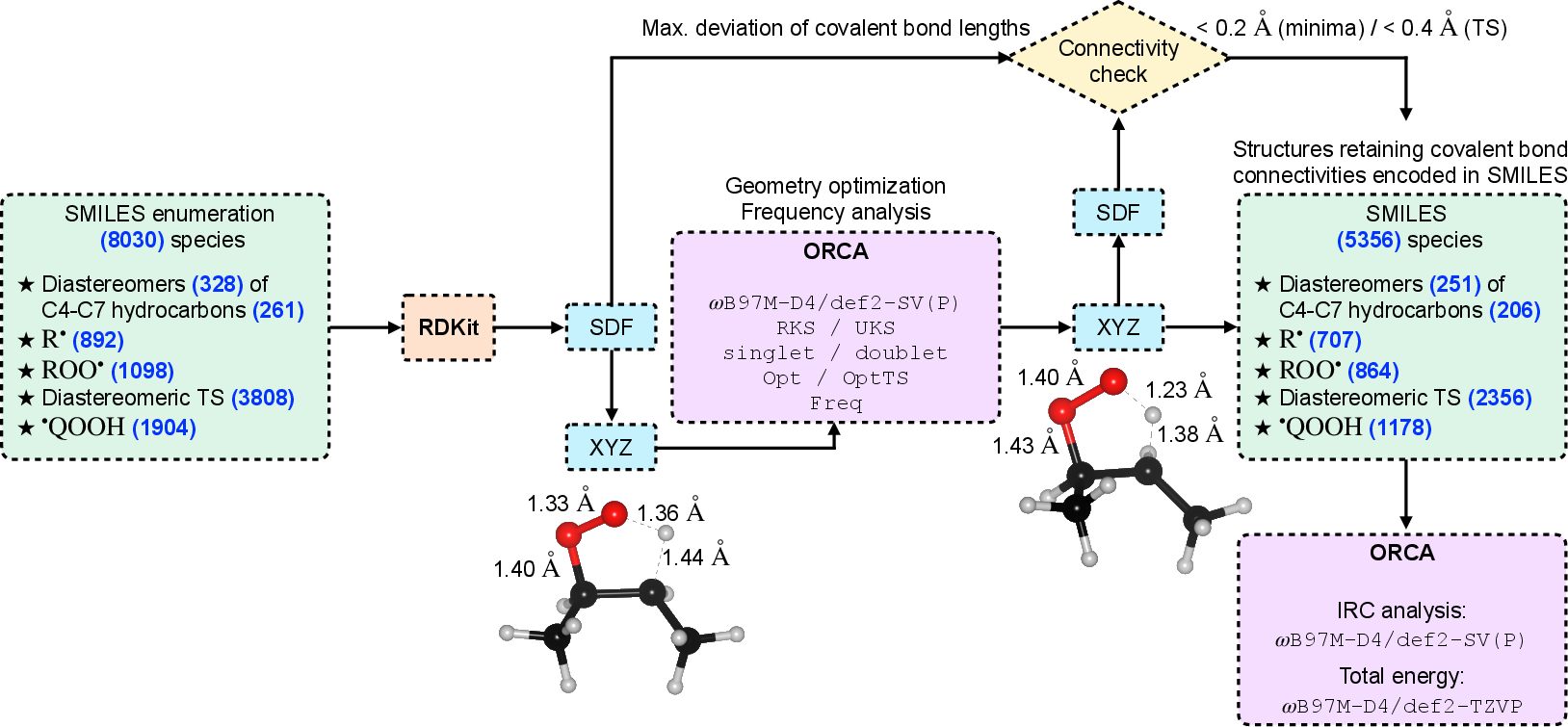
Figure 2: Quantum chemistry workflow for generating, optimizing, and validating diastereomeric transition states for ROO∙→∙QOOH reactions.
Out of 3,808 initial candidate transition states, 2,324 structures (1,162 diastereomeric pairs) were verified to connect the correct minima, forming the core dataset.
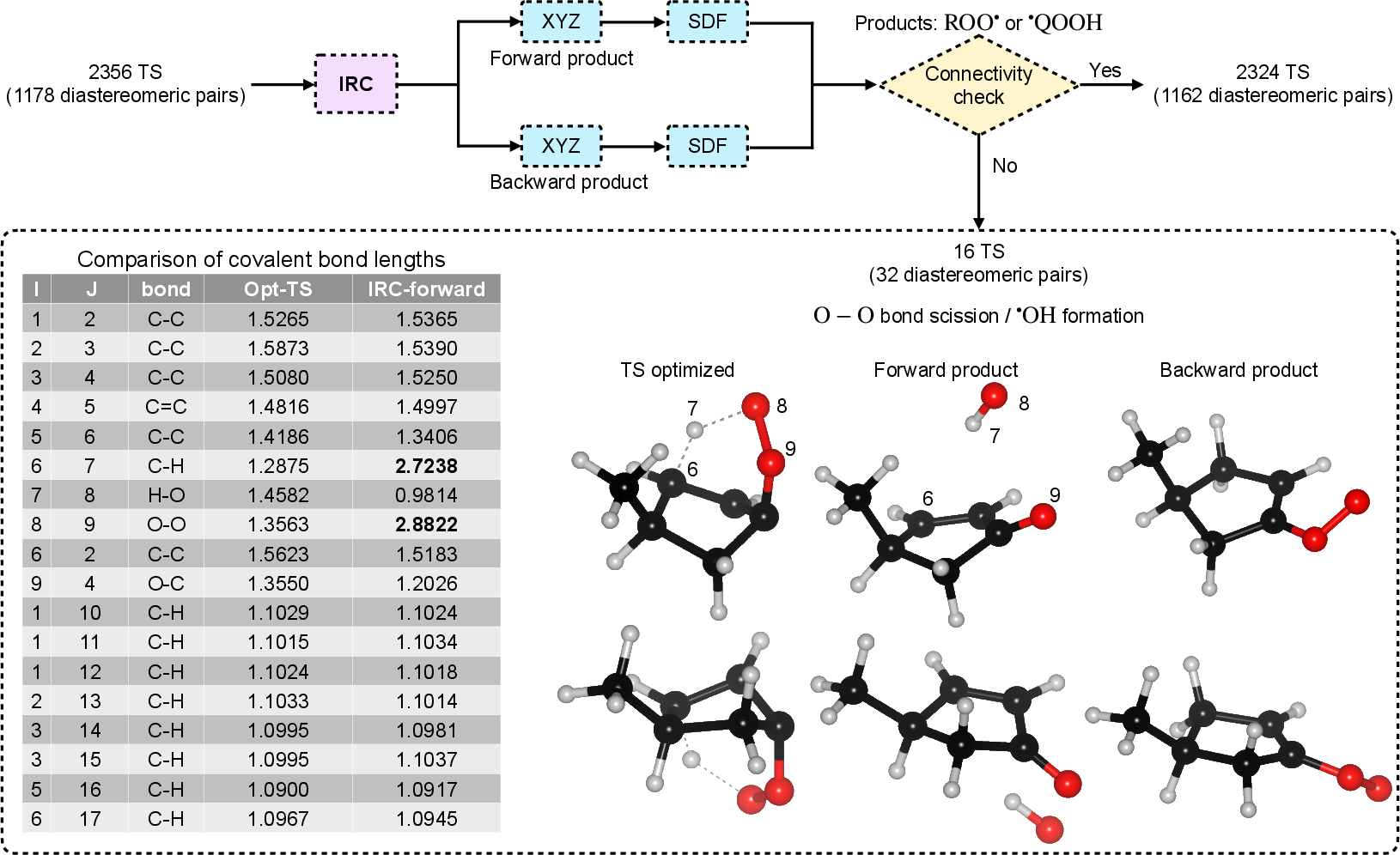
Figure 3: IRC analysis workflow illustrating connectivity validation for 2,356 transition-state candidates, discriminating between intended H-shift and off-path processes such as O–O scission.
Energetic Landscape of Diastereomeric Barriers
The joint distribution of reaction energies and forward barrier heights (ΔEbarrier) across all validated transition states underscores the nontrivial stereochemical diversity in the barrier landscape. Each reaction yields a pair of diastereomeric transition states, typically degenerate in reaction energy but frequently distinct in barrier height. The magnitude of the barrier splitting, ΔΔEbarrier=ΔEhigh−ΔElow, was found to range from sub-kcal/mol to over 60 kcal/mol, with the majority of pairs exhibiting ΔΔEbarrier≤3 kcal/mol.
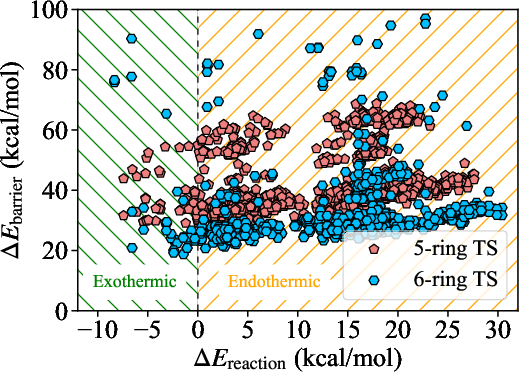
Figure 4: Scatter plot of reaction energies vs forward barrier heights for 1,162 diastereomeric pairs; each reaction displays two points horizontally aligned (identical ΔErxn) but with potentially distinct C10.
The distribution of these energy differences is detailed in a histogram highlighting that 32% of pairs are nearly degenerate (C11 kcal/mol), but a nontrivial fraction possess substantial splittings, indicating one pathway may be kinetically dominant while the other is irrelevant.
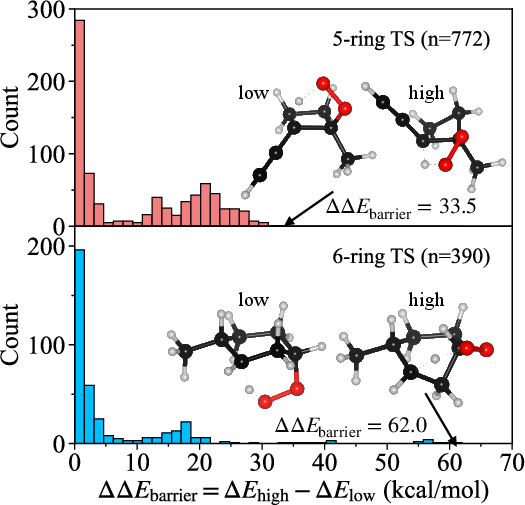
Figure 5: Histogram of C12 values for all diastereomeric pairs, illustrating the prevalence of near-degeneracies but also a significant tail of large splittings.
Structural analysis by transition-state ring size, peroxyl-bearing carbon hybridization, and H-abstraction site substitution reveals systematic trends: large splittings are most pronounced when the peroxyl group is at an C13 center and the H-abstraction site is secondary, especially in cyclic or sterically constrained systems.
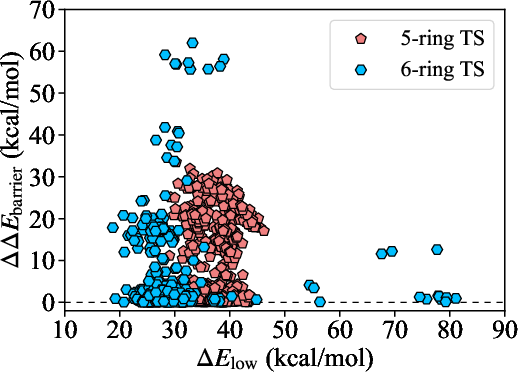
Figure 6: Scatter plot of diastereomeric energy splitting vs lower barrier, resolving trends by structural features such as ring size and substitution.
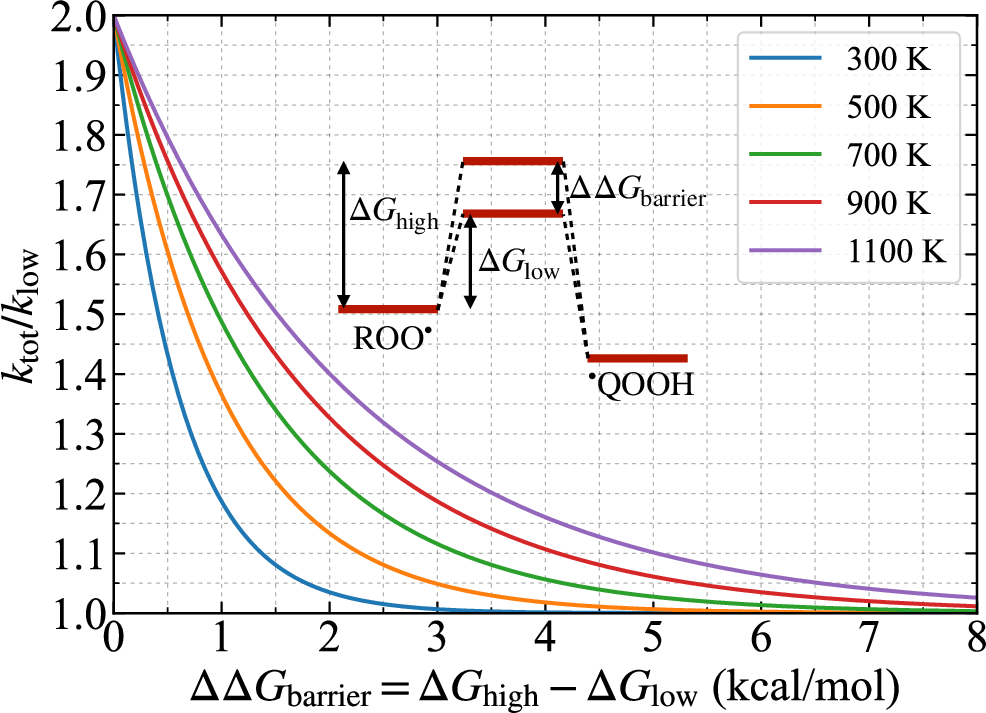
Figure 7: Kinetic consequence of parallel diastereomeric pathways—rate enhancement factor vs barrier splitting, at multiple temperatures.
Implications for Reaction Kinetics and Mechanism Generation
The kinetic impact of diastereomeric barrier splitting is derived from transition-state theory. For reaction pathways with nearly degenerate diastereomeric barriers, the rate constant is essentially doubled relative to a single pathway. However, even a moderate splitting (e.g., C14 kcal/mol) renders the higher barrier pathway kinetically negligible at combustion-relevant temperatures, leading to significant undercounting of reaction rates in constitutionally collapsed mechanism representations.
The data indicate that such splitting is a common feature in hydrocarbon autooxidation, especially for conformationally inflexible molecular classes where ring strain or substituent orientation penalizes one diastereomeric transition state.
These findings underscore a methodological deficiency in current high-throughput or automated mechanism generation pipelines, which frequently fail to propagate and track stereochemical distinctions at the transition-state level. The omission of diastereomeric transition states can result in the systematic undercounting of kinetically relevant channels, impacting both rate estimation and the identification of dominant pathways.
Representative Reaction Profiles
Detailed examination of representative exothermic C15 reactions corroborates the theoretical analysis: in some cases, diastereomeric barriers are nearly identical; in others, barrier splitting is pronounced, and only one channel contributes.
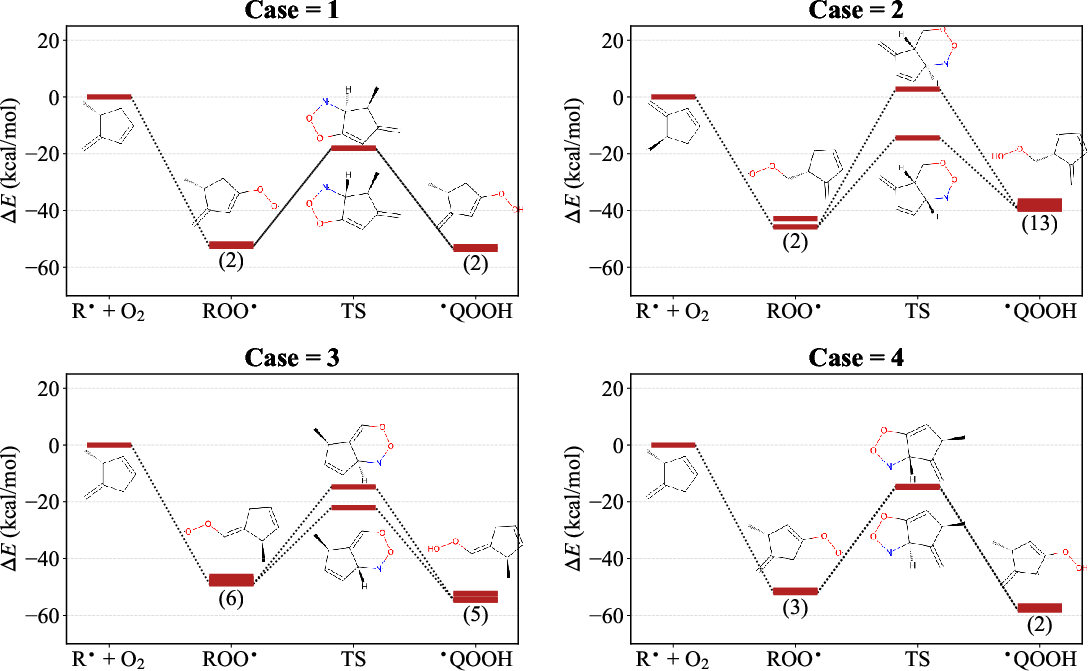
Figure 8: Energy profiles for selected reactions, with conformational isomers of reactants, two diastereomeric transition states, and product conformers; transition-state structures and splitting explicitly visualized.
These specific analyses reinforce the system-dependent nature of stereochemical effects: ring size, local hybridization, and substitution patterns co-determine the barrier landscape.
Perspectives and Future Developments
The SEARS dataset and associated stereochemical enumeration protocols provide a scalable framework for stereochemistry-aware generation of reaction networks. Incorporation of these protocols into automated combustion modeling platforms will improve accuracy, especially for molecules where multi-channel, non-degenerate diastereomeric H-shifts are prevalent.
The data offer a challenging benchmark for machine learning models intended to predict reaction barriers and rate constants, emphasizing the need for molecular representations and architectures competent in capturing subtle stereoelectronic and stereochemical nuances beyond constitutional topology. Graph-based formalisms such as StereoMolGraph are poised to facilitate integration of stereochemistry at scale in computational reaction discovery (2604.17357).
Conclusion
This comprehensive analysis elucidates the critical influence of transition-state stereochemistry on autooxidation kinetics of small hydrocarbons. Diastereomeric transition states are not exceptions but a prevalent feature, with barrier splitting routinely exceeding the threshold for kinetic relevance. The explicit treatment of stereochemistry in both data and workflow enables more accurate mechanism development and predictive modeling for combustion, atmospheric, and more general radical-mediated oxidative chemistries. The principles, dataset, and computational protocols established here will inform both fundamental studies and the continued development of next-generation data-driven kinetic modeling tools.