- The paper introduces LCCVOs to construct localized virtual orbitals from PW-DFT, significantly reducing basis size while improving correlation energy recovery.
- The paper demonstrates that LCCVOs yield superior dissociation energy accuracy across closed-shell, doublet, and triplet molecules relative to traditional atom-centered bases.
- The paper shows that LCCVOs enable efficient quantum algorithm integration by mapping to reduced Hamiltonians, thereby lowering qubit and circuit complexity.
Localized Correlation-Converged Virtual Orbitals for Quantum Molecular Simulations
Introduction
The accurate treatment of electron correlation is a central challenge in quantum chemistry, especially as quantum hardware and advanced many-body methods become more relevant for simulation of molecular systems. Conventional approaches based on Gaussian-type orbitals (GTOs) offer systematic convergence but scale poorly with the number of basis functions, impeding their integration with near-term quantum devices. Plane-wave (PW) basis sets, while providing several algorithmic advantages and systematically improvable completeness, suffer from severe deficiencies in the description of virtual (unoccupied) orbitals—most notably, delocalization into vacuum states that deteriorate correlation energy recovery. The work introduces Localized Correlation-Converged Virtual Orbitals (LCCVOs): a framework for constructing efficient, compact virtual orbital spaces from PW-based DFT, for use in post-mean-field correlated methods and, crucially, scalable quantum algorithms (2604.16928).
Theoretical Framework
The LCCVO approach is motivated by the inadequacy of conventional virtual orbitals generated via DFT when using PW expansions, which typically leads to the inclusion of delocalized and unphysical vacuum states in the active space of many-body calculations. These artifacts diminish the effectiveness of correlated methods (e.g., CCSD) and undermine the accuracy of quantities such as molecular dissociation energies.
The LCCVO strategy eliminates these defects by:
- Starting from Kohn-Sham orbitals obtained via a PW-DFT calculation subject to a finite simulation cell.
- Ranking and filtering the virtual orbitals such that only those contributing significant correlation energy (as measured by CCSD) are retained.
- Iteratively optimizing each virtual orbital for correlation, ensuring locality and strong coupling to the physically relevant occupied space.
- Exclusion of pure and mixed vacuum states based on their negligible impact on correlation energy recovery.
By aligning the virtual orbital space closely to the occupied manifold and local physical structure, the LCCVO protocol constructs minimal, high-fidelity Hamiltonians.
Numerical Benchmarks
Molecular test cases span closed-shell (\ce{H2}, \ce{N2}, \ce{C2}), doublet (\ce{CN}), and triplet (\ce{O2}) species, encompassing both weak and strong correlation regimes and open-shell electronic structures. Compared to high-level atom-centered basis sets (cc-pVXZ, X=D–5), the LCCVO description delivers three principal advantages:
- Superior correlation energy recovery with compact basis: For all molecules except \ce{H2}, LCCVOs achieve lower errors in dissociation energy relative to the largest conventional basis sets, using only 15–50 orbitals (vs. 110–182 for cc-pV5Z).
- Broad state applicability: Unlike previous approaches (e.g., COVO, limited to closed shells), LCCVO directly extends to doublet/triplet and complex multi-reference systems.
- Improved agreement with experiment: In several cases (notably \ce{N2}, \ce{O2}, \ce{CN}), the error in dissociation energy falls below the cc-pV5Z and even surpasses the CBS extrapolated limit.
The total energy as a function of interatomic separation for prototypical closed-shell and open-shell molecules demonstrates that the LCCVO methodology robustly captures the correct dissociation behavior, even with a greatly reduced virtual space.
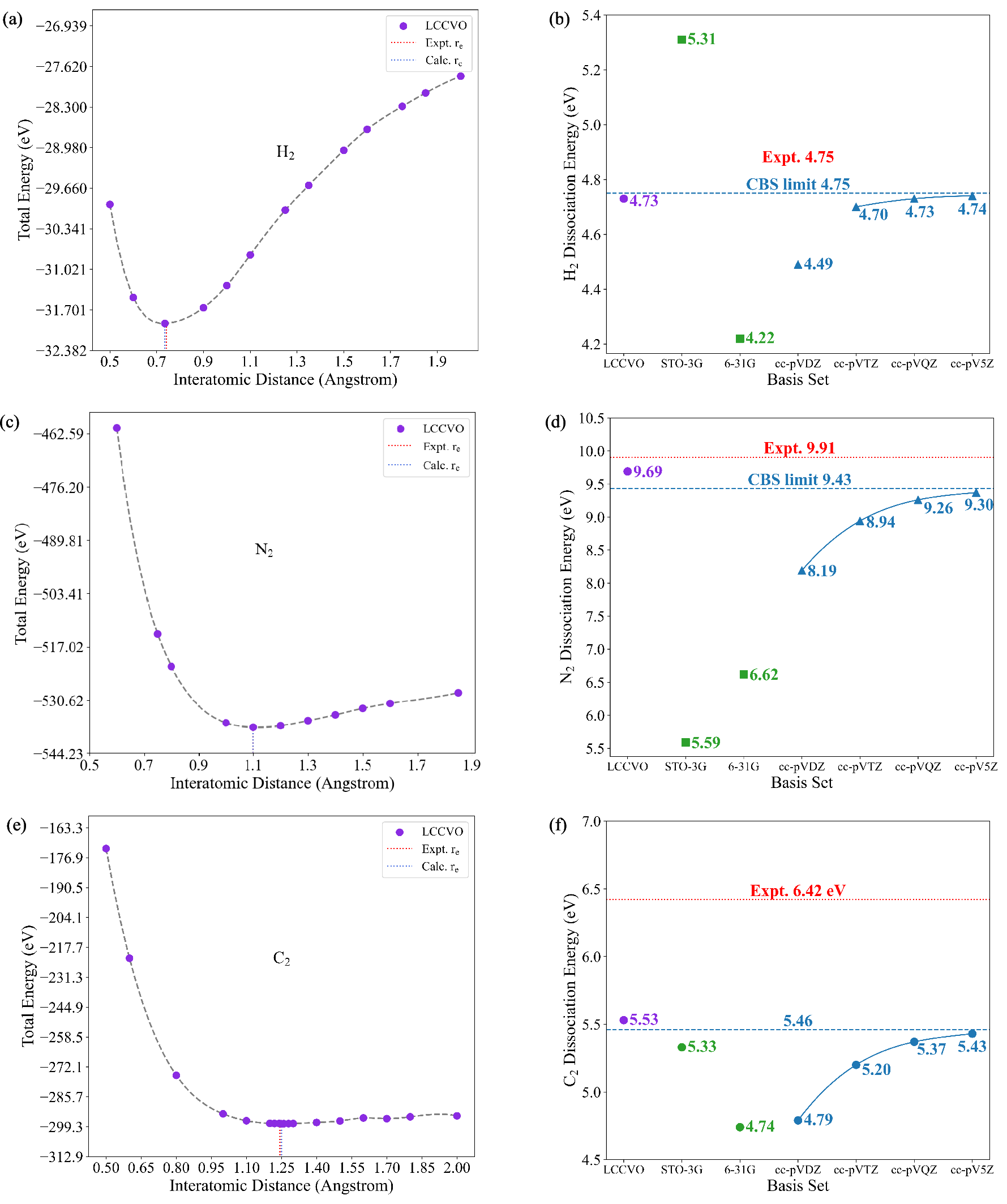
Figure 1: LCCVO achieves high-fidelity total energies for singlet states while requiring far fewer virtual orbitals than atom-centered approaches.
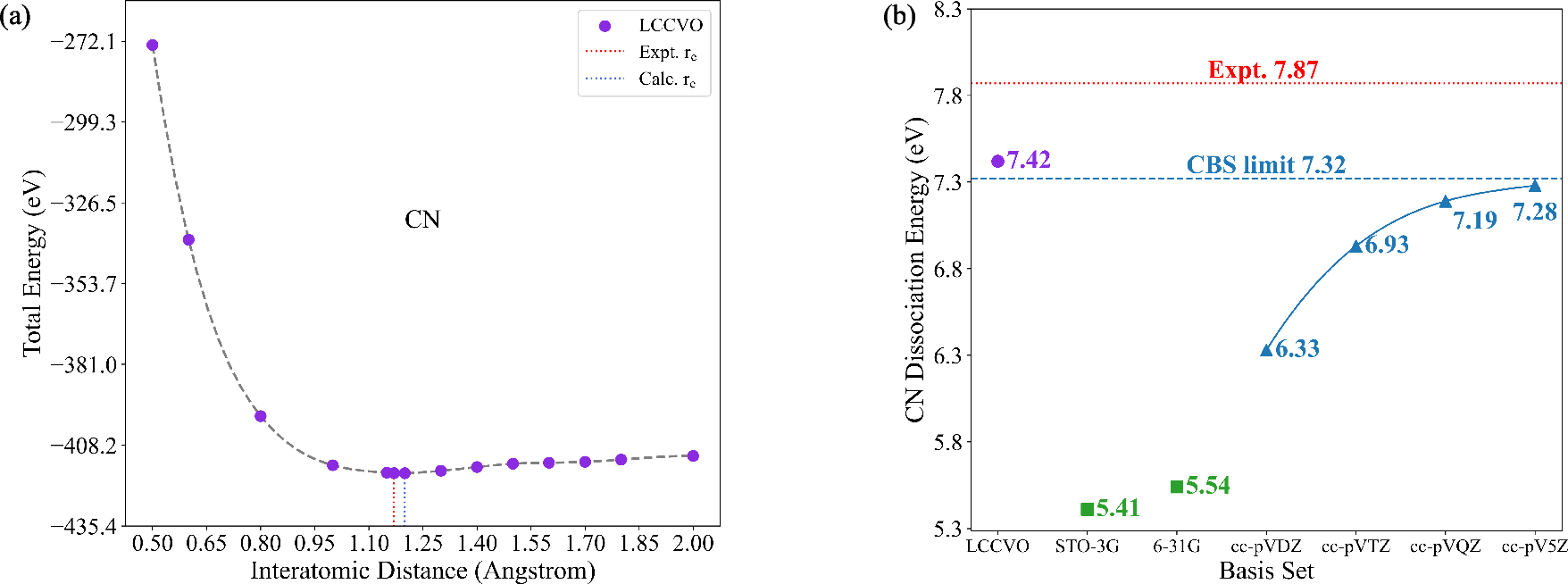
Figure 2: LCCVO captures the entirety of the dissociation curve for open-shell doublet systems such as \ce{CN} with quantitative accuracy.
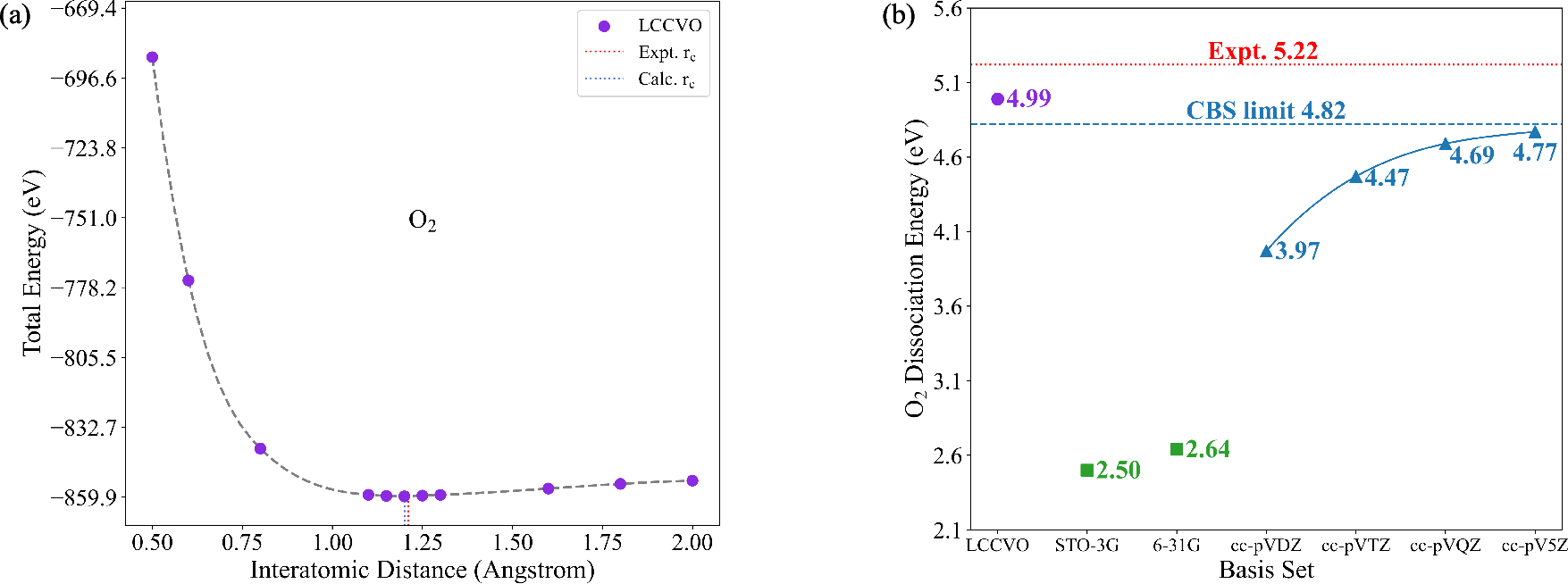
Figure 3: For triplet systems like \ce{O2}, the LCCVO method demonstrates superior accuracy and orbital economy compared to large GTO bases.
Furthermore, spatial visualization confirms that LCCVOs eliminate unphysical vacuum delocalization present in standard PW-DFT orbitals, localizing the relevant virtual space on the molecule itself. This property is essential for achieving efficient and accurate post-mean-field correlation calculations.
Correlation Energy Recovery and Algorithmic Scaling
Direct comparison of correlation energy recovered with and without LCCVO demonstrates the critical advantages of the proposed protocol in N2 and related systems:
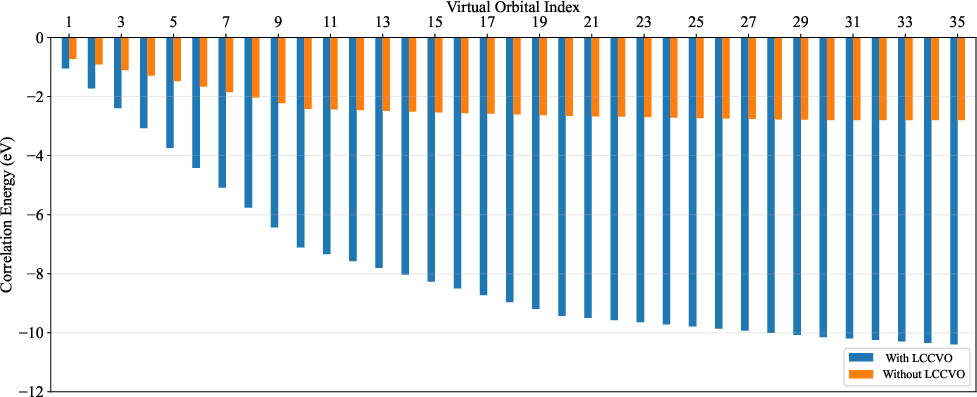
Figure 4: LCCVOs extract substantially more correlation energy for a given number of virtual orbitals as compared to conventional KS orbitals.
The O(1/N) error scaling with respect to the total number of included LCCVOs mirrors high-level atom-centered basis set convergence, yet with a steeper slope reflecting more efficient orbital utilization. CCSD total energies converge smoothly with expansion of the LCCVO space, and systematic extrapolation protocols with respect to both orbital number and vacuum cell length mitigate finite-size PW artifacts.
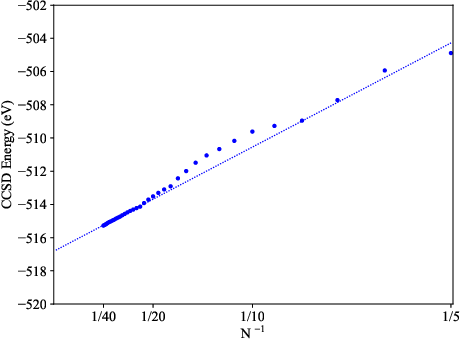
Figure 5: The CCSD energy for N2 converges rapidly as a function of the reciprocal number of LCCVOs, validating minimal-basis flexibility.
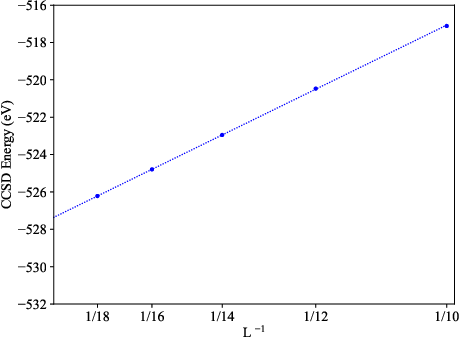
Figure 6: CCSD total energies exhibit expected O(1/L) scaling with increasing simulation cell, demonstrating reliable finite-size correction via extrapolation.
Discussion and Implications
This work establishes that basis set selection—and, specifically, the construction of correlation-optimized virtual locality—remains a pivotal issue for both classical and quantum post-DFT electronic structure. LCCVOs resolve the principal limitation of the PW expansion for isolated non-periodic systems: the rapid growth and unphysical character of the unoccupied manifold.
Strong quantitative claims include:
- LCCVO yields errors in dissociation energies for multi-electron (e.g., \ce{N2}) and open-shell (e.g., \ce{O2}, \ce{CN}) systems that are below or comparable to large-basis (cc-pV5Z, CBS) reference values using 3×–10× fewer orbitals.
- In some cases, LCCVO energies are more accurate than CBS extrapolations.
The formalism is directly compatible with quantum algorithms predicated on second quantization, where the number of qubits and circuit complexity scales linearly with the number of spatial orbitals. Thus, LCCVOs dramatically lower the hardware and computational threshold for meaningful quantum simulation of correlated molecular systems.
For the problematic \ce{C2} case, residual error (>10%) persists, attributable also to unresolved experimental ambiguities and not solely to method limitations, as detailed in the supplementary analysis.
Future Directions
Potential avenues for development include:
- Extension to periodic/solid-state systems: LCCVO construction with explicit k-point sampling is straightforward and may supplant inefficient GTOs or raw PWs for correlated solids.
- Integration with higher-excitation methods: Inclusion of CCSDT or FCI within the LCCVO framework is anticipated to close any remaining gaps to experimental observables.
- Synergy with quantum algorithms: Direct mapping of LCCVO-truncated Hamiltonians to variational quantum eigensolver (VQE), Quantum Phase Estimation (QPE), or related hybrid protocols may accelerate practical quantum chemistry on early fault-tolerant hardware.
Conclusion
LCCVOs deliver a robust, physically motivated basis for efficient and accurate quantum molecular simulation, particularly for strongly correlated and open-shell species. By optimally localizing and truncating the virtual orbital space, LCCVOs enable high-accuracy energetics with an order-of-magnitude fewer basis functions than traditional GTO approaches—an essential capability for both classical and quantum correlated methods (2604.16928). This framework lays a foundation for expanded chemical application on near-term quantum hardware and establishes a blueprint for systematic basis construction beyond the limitations of atom-centered or raw plane-wave schemes.

