- The paper presents a novel QUBO formulation that integrates explicit Coulomb, van der Waals, hydrogen bonding, and hydrophobic interactions for molecular docking.
- The method encodes both ligand and protein pockets as augmented graphs with atomic-level features, enabling accurate subgraph isomorphism mapping.
- Experimental results show a 20% reduction in mean Adjusted RMSD and sub-angstrom accuracy, while highlighting current quantum hardware constraints.
Introduction
Molecular docking underpins high-throughput structure-based drug design, with computational demands stemming both from the combinatorial pose search and the complexity of the energetics underlying the protein-ligand interface. Quantum annealing has emerged as a classically-intractable candidate for driving combinatorial optimization, but prior quantum annealing applications to molecular docking were limited by constraints of expressivity. Specifically, they relied primarily on geometric compatibility and lacked explicit representations of critical physico-chemical interactions at the atomic level. This work presents a novel Quadratic Unconstrained Binary Optimization (QUBO) formulation embedding Coulomb, van der Waals (vdW), hydrogen bond (H-bond), and hydrophobic interactions, enabling more physically faithful molecular docking on quantum hardware (2604.09540).
Methodological Innovations
Graph-Based Representation with Physicochemical Augmentation
Both the ligand and protein pocket are encoded as graphs: Gmol (ligand) and Ggrid (pocket), with vertices representing atoms and discretized grid points, respectively. Beyond previous geometric models, this work augments nodes and edges with additional node-specific features capturing partial charges, vdW parameters, H-bond donor/acceptor roles, and hydrophobicity. These are precomputed for all relevant atom types using established force fields (e.g., MMFF94) and empirical geometric criteria for interaction potential.
The ligand-to-pocket embedding problem is formulated as a weighted subgraph isomorphism, converted to QUBO via indicator variables xij denoting the mapping of ligand atom i to grid node j. The QUBO objective is:
H=Hgeom+∑kλkHk
where Hgeom encodes geometric compatibility and injectivity, and each Hk encodes contributions from one physical interaction:
- Coulomb: Hel encodes atom charge (qi) mapped to precomputed electrostatic potential (Ggrid0).
- van der Waals: Ggrid1 comprises precomputed Lennard-Jones energies parameterized for atom types.
- H-bonds: Ggrid2 and Ggrid3 model donor/acceptor feasibility via geometric and role criteria.
- Hydrophobic: Ggrid4 penalizes/exploits matching hydrophobic regions.
All terms are kept quadratic, resolving a key expressivity challenge for QUBO and avoiding the variable proliferation of HUBO-to-QUBO reductions.
Experimental Design
Dataset and Metrics
The experimental evaluation utilized subsets of PDBbind 2020, restricting ligand size to enable tractable quantum hardware mapping (Ggrid5 heavy atoms for simulated annealing, Ggrid6 for D-Wave). Docking quality was assessed via "Adjusted RMSD," compensating for grid discretization by subtracting the lowest achievable RMSD given the grid resolution from the predicted pose RMSD.
Hyperparameter Optimization
A greedy approach selected optimal Ggrid7 coefficients for the QUBO, revealing the relative importance of physical interactions. The geometric weight was fixed, and interaction terms were incrementally tuned to minimize mean Adjusted RMSD over complexes, showing van der Waals and Coulomb forces dominate, with non-negligible contributions from hydrophobic and H-bond (acceptor) terms.
Results and Analysis
Simulated annealing demonstrates a 20% reduction in mean Adjusted RMSD for the proposed physically-informed QUBO compared to the geometric baseline. Variance is also reduced, indicating greater reliability in pose prediction.
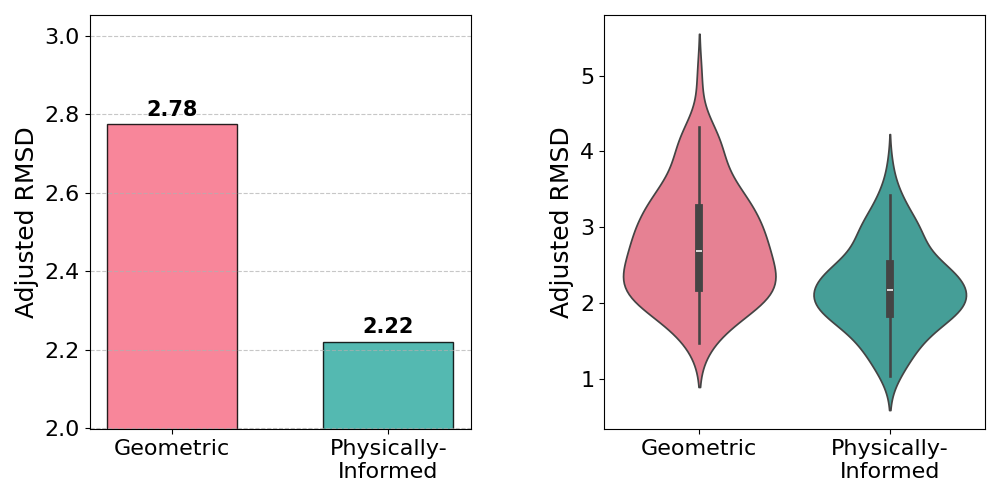
Figure 1: Comparison of the fully geometric approach and the proposed physically-informed method using Simulated Annealing, highlighting improvements in docking accuracy and consistency.
On D-Wave Advantage, the mean Adjusted RMSD improvement exceeds 15% despite more restrictive problem sizes. Furthermore, some predicted poses approach sub-angstrom accuracy relative to experimental structures.
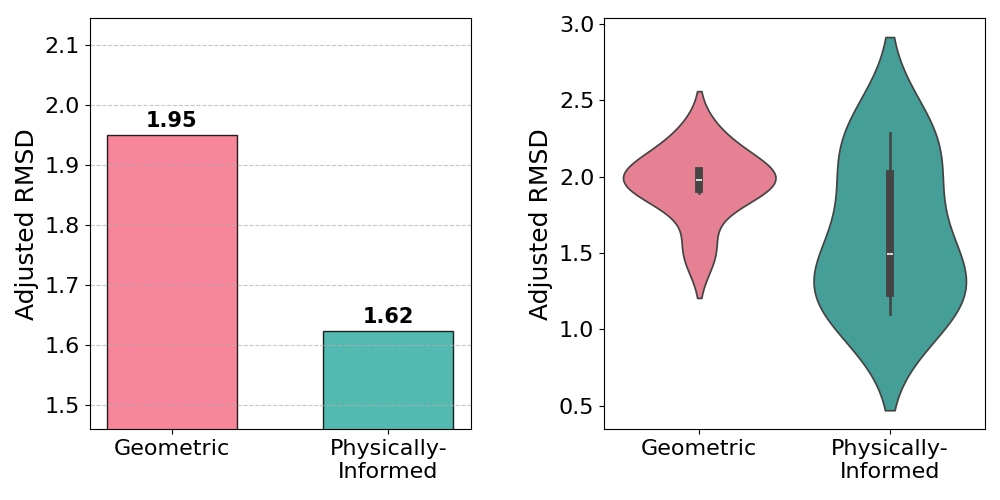
Figure 2: Comparison of the fully geometric approach and the proposed method on D-Wave Advantage, highlighting improved docking accuracy.
However, the absolute yield of valid solutions (embedding success rates) on current QPU hardware is extremely low (sub-1%), even after extensive exploration of chain strength, annealing time, and embedding algorithms. The physical qubit overhead is an order of magnitude above logical variables, underlining the need for improved encoding and mapping strategies.
Characterization of Interaction Relevance
The hyperparameter optimization sets the van der Waals and Coulomb terms to the highest weights, confirming these interactions are most influential in accurate binding pose determination. H-bond and hydrophobic terms, while weaker contributors at the global level, are essential for specific complexes where such interactions dominate binding specificity. The empirical absence of a donor H-bond contribution (Ggrid8) indicates interaction context may override purely chemical potential, potentially due to limitations in the input models or dataset distribution.
Quantum Annealing Hardware Constraints
Both DWaveSampler and DWaveCliqueSampler embeddings exhibit high physical-to-logical qubit overhead and long average chain lengths. Despite the high connectivity of the Pegasus topology, current quantum annealers cannot efficiently scale to biologically relevant complex ligands. Adjustments in hardware parameters failed to appreciably increase valid solution yield, indicating a systemic limitation of present hardware and QUBO mapping.
Implications and Future Directions
This work establishes the feasibility and measurable benefit of physically-informed docking formulations for quantum annealing. The ability to encode van der Waals, Coulomb, H-bond, and hydrophobic interactions simultaneously, with all terms natively quadratic, closes a critical gap between physically consistent scoring functions and QUBO-compatible quantum hardware. However, technological bottlenecks in quantum hardware embedding, problem scaling, and solution yield must be overcome for the approach to be applicable to large-scale drug discovery.
Looking forward, progress is anticipated in:
- Encoding efficiency: Exploring domain-wall or other compact encodings to reduce physical qubit overhead, as suggested in the original work.
- Scalability: Generalizing the QUBO formulation to multi-ligand and fragment-based docking, although the variable count presents new computational challenges.
- Hardware maturation: Advancements in QPU connectivity, logical qubit density, and error correction will be required for practical adoption in CADD pipelines.
- Fine-tuned interaction modeling: Selective inclusion or context-dependent weighting of physicochemical terms may yield further accuracy improvements as datasets and models become more comprehensive.
Conclusion
The integration of physically realistic interaction terms into a graph-based QUBO framework for molecular docking successfully bridges the gap between geometric matching and atomistic physical realism within the constraints of quantum annealing architectures. The observed improvements in docking accuracy validated both in classical and quantum annealing settings highlight the direction for future quantum approaches in drug discovery. Yet, utilization at scale will depend on parallel progress in encoding strategies and quantum hardware capabilities.