- The paper presents an open-source Python framework that implements the anti-Hermitian contracted Schrödinger equation for precise all-electron correlation.
- It employs iterative RDM minimization with 3-RDM reconstruction (Valdemoro and NY), achieving superior benchmarks compared to NEVPT2 and FCI methods.
- The method efficiently computes both ground and excited states in strongly correlated regimes, proving effective for transition metal and main group systems.
Open-source Implementation of the Anti-Hermitian Contracted Schrödinger Equation for Multistate All-electron Correlation
Introduction and Theoretical Foundation
This work provides a systematic presentation and benchmark of an open-source Python implementation of the anti-Hermitian contracted Schrödinger equation (ACSE) for electronic ground and excited states (2604.02550). The effort addresses critical challenges inherent in the ab initio simulation of strongly correlated electrons—specifically, the accurate treatment of all-electron correlation in the context of chemically and technologically relevant systems, where standard configuration interaction (CI)-based and multireference perturbation approaches are computationally prohibitive or suffer from significant limitations.
The ACSE formalism operates entirely in the language of the reduced density matrix (RDM) hierarchy. Specifically, the solution targets the anti-Hermitian component of the contracted Schrödinger equation, written in terms of the 2-RDM with approximate reconstruction of contributions from the 3-RDM, thereby enabling efficient, wavefunction-free computation of many-electron correlation effects. In contrast to many widely used alternatives (e.g., NEVPT2 or MRPT), the ACSE operates independent of the complexity of the reference wavefunction and utilizes the exact electronic Hamiltonian without introducing an effective Hamiltonian or associated artifacts such as intruder states. The computational scaling is O(r6) in the number of molecular orbitals, with memory scaling as O(r4).
The ACSE is solved via iterative minimization of the RDM residual, informed by gradient steps, with specific attention paid to the maintenance of N-representability of the evolving RDM and the effect of approximations inherent to various 3-RDM reconstruction strategies.
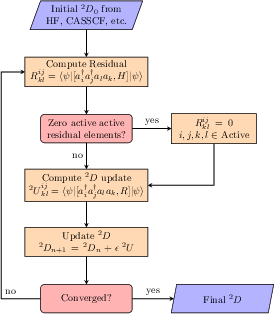
Figure 1: Overview of the implemented ACSE computational workflow, highlighting Python, PySCF interface, and tensor contraction scheme.
Benchmarking Methodology and Systems
A broad suite of benchmarks emphasizes representativity and rigor, encompassing main group and transition metal systems, weakly and strongly correlated regimes, and ground as well as excited states. Comparisons are made systematically against both NEVPT2 (PySCF implementation) and FCI or DMRG-FCI reference energies. Basis sets span 6-31G, cc-pVDZ, cc-pVTZ, def2-SVP, and def2-TZVP, and active spaces range from minimal ([2,2]) to extended ([8,8]), with all calculations utilizing either Hartree-Fock or state-averaged CASSCF references. The two most established 3-RDM reconstruction approaches are supported: Valdemoro (V) and Nakatsuji-Yasuda (NY). Active-active propagation protocols are examined to systematically investigate their effect on accuracy and convergence.
Results: Ground-State and Excited-State Energetics
H\$_6\$ Dissociation and Dihedral Barriers
For symmetric dissociation of linear H6, the ACSE results are generally more accurate than NEVPT2, with the NY reconstruction preferred for cases where active-active propagation is permitted, and the V functional more robust when this is disabled. Notably, the error in ACSE remains stable with respect to increasing basis set size, whereas NEVPT2 degrades (MSE increases from 2.34 to 11.56 mH across 6-31G to cc-pVTZ), highlighting inherent basis set compatibility in the ACSE approach.
In the context of ethylene (C2H4) dihedral rotation, the ACSE displays an order of magnitude superior accuracy in absolute energies compared to NEVPT2: maximum barrier error ≈0.54 kcal/mol with the V (False) protocol, versus ≈1.35 kcal/mol for NEVPT2.
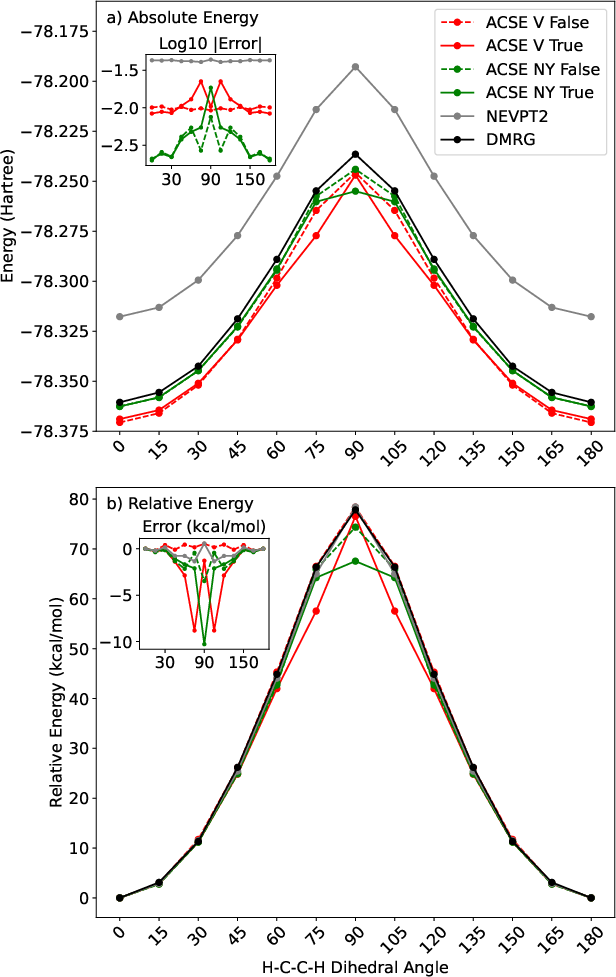
Figure 2: Absolute and relative energies for ethylene dihedral barrier—ACSE (V and NY reconstructions, with/without active-active propagation) and NEVPT2 against DMRG-FCI reference.
Convergence analysis indicates the V reconstruction is stable in both weak and strong static correlation regimes; the NY approximation, however, often fails to converge and yields large errors under strong correlation, consistent with its derivation under a single-reference paradigm.
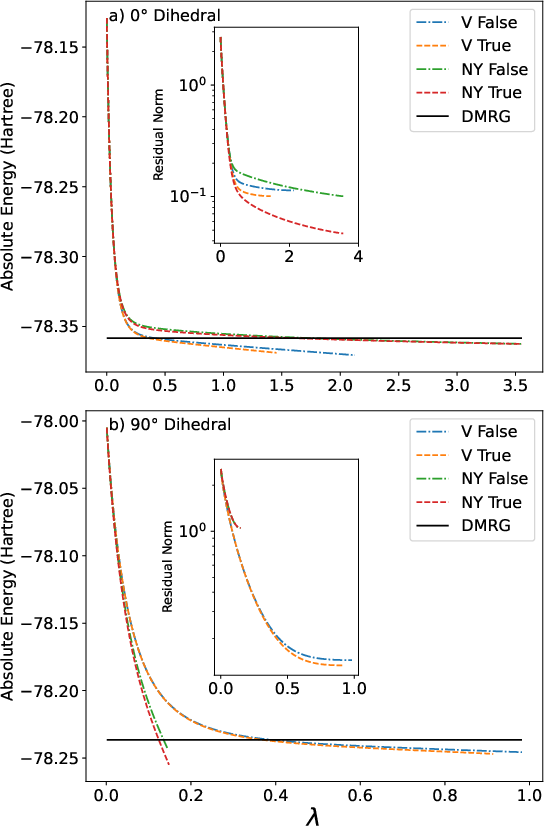
Figure 3: Energy and residual norm convergence for ethylene at 0° and 90° dihedral, revealing qualitative differences between V and NY reconstructions.
Excited-State Calculations
For vertical excitations, specifically the S0→ S1 transition in ethylene, the ACSE (V, False) achieves average errors less than 10 meV using the [2,2] active space, outperforming NEVPT2 (average error O(r4)0250 meV). However, as the active space increases, the error profile in ACSE becomes less favorable due to neglected higher-order cumulants in the V reconstruction. The NY approach again performs poorly when significant multireference character emerges.
The robustness of the ACSE is further demonstrated by atomic spin splittings in FeO(r4)1, CoO(r4)2, and FeO(r4)3. For FeO(r4)4, ACSE (def2-TZVP) errors are reduced to 1.47, 2.14, and 0.63 kcal/mol (triplet-singlet, quintet-singlet, quintet-triplet) compared to experimental benchmarks, which is competitive with or superior to NEVPT2 and substantially better than CASSCF. This trend is preserved for CoO(r4)5 and FeO(r4)6, establishing the capability of the ACSE to resolve spin energetics in open-shell, transition-metal-rich environments.
NO(r4)7 Dissociation and State Ordering
Along the NO(r4)8 dissociation coordinate, ACSE (V, False) provides maximal errors O(r4)91 kcal/mol near minima in SN0, TN1, and TN2, growing to 5–7 kcal/mol at larger bond lengths. This behavior remains superior to NEVPT2 for absolute correlation energies but both methods track state ordering and qualitative PES features consistently.
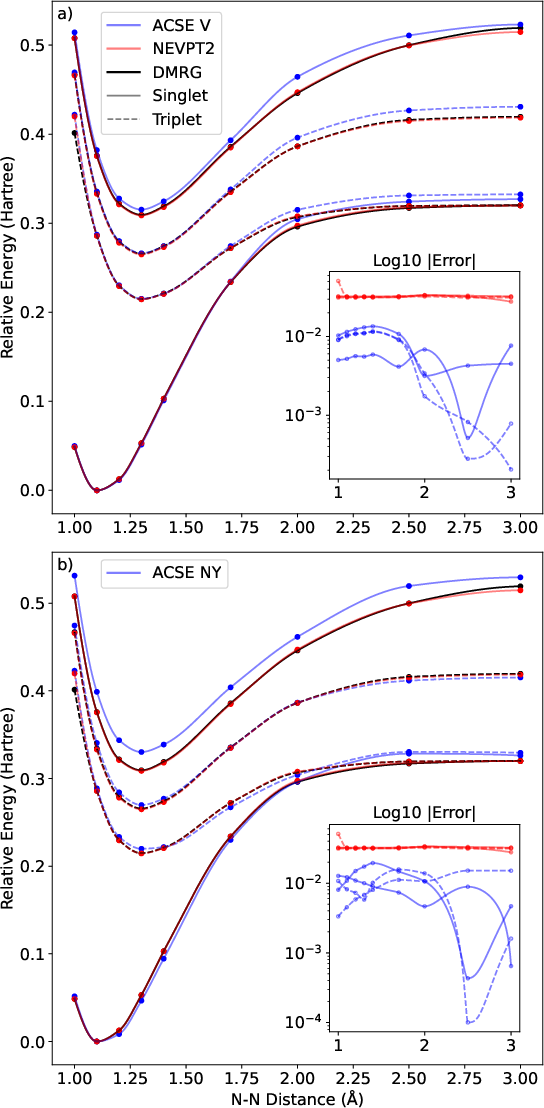
Figure 4: Ground and excited, singlet and triplet potential energy surfaces for NN3 dissociation—ACSE (V and NY reconstructions) and NEVPT2 versus DMRG-FCI. Insets: log of absolute error to DMRG-FCI.
Algorithmic, Practical, and Theoretical Implications
This work establishes the open-source ACSE as a scalable, robust alternative to established multireference electron correlation approaches, with several distinctive properties:
- Hamiltonian fidelity: The ACSE operates with the exact (non-perturbative) Hamiltonian and does not require an effective Hamiltonian or approximate treatment which can introduce artifacts (e.g., intruder states or surface discontinuities).
- Active space independence: Computational scaling is not a function of the reference wavefunction’s multireference complexity, offering clear advantages for large active-space scenarios.
- Multistate direct access: Both ground and targeted excited states are directly accessible by appropriately selected reference RDMs, simplifying state-specific treatments and root flipping.
- Open-source accessibility: The modular, python-based implementation with seamless PySCF interoperability accelerates adoption and extension, facilitating integration with modern electronic structure packages.
However, the results demonstrate that reliable ACSE performance is contingent upon judicious choice of 3-RDM reconstruction (favoring V in strongly correlated regimes, while NY suffers in multiconfigurational contexts) and careful residual propagation without excessive activation of active-active elements, which can otherwise destabilize the minimization or yield unphysical RDMs.
Prospects for Future Development
Outstanding challenges relate to systematically improving 3-RDM reconstruction—specifically, the need for robust, general-purpose functionals suitable for highly multireference states. Incorporation of the Mazziotti functional is proposed as a particularly promising direction. Additional algorithmic accelerations (symmetry exploitation, parallelization) and convergence strategies (asymptotic fitting, smart extrapolation) are anticipated to further improve stability and applicability.
Potential applications extend to extended strongly correlated systems, excited-state PES mapping, transition-metal cluster catalysis, and the exploration of open-shell transition-metal chemistry—situations where current multireference methods quickly become intractable.
Conclusion
The presented open-source ACSE implementation marks a substantial advance in making high-fidelity, all-electron correlated simulations tractable for a broad class of chemical systems, including ground and excited states in main group and transition metal chemistry. The methodology offers compelling accuracy, particularly in absolute energy and spin state ordering, with efficient scaling and Hamiltonian fidelity. Current best practice dictates the use of the Valdemoro reconstruction without active-active residual propagation for strongly correlated states; further algorithmic advances in the area of 3-RDM reconstruction will be pivotal for systematic deployment to even larger systems and more complex multistate environments.

