- The paper introduces a novel automated pipeline that uses external embryoid body controls and background subtraction to recover TF-specific transcriptional signatures despite incomplete controls.
- It accurately assigns cells to 87 TFs and doubles the detected differential expression compared to naive methods, ensuring robust analysis across 30 experiments.
- The study validates its findings by benchmarking against published scRNA-seq rankings, demonstrating effective artifact correction and minimal batch effects for reliable TF regulatory insights.
Introduction
This study presents a comprehensive re-analysis of the Human Transcription Factor (TF) Atlas dataset (GSE216481), originally generated by Joung et al. using the MORF (Mammalian ORF) pooled lentiviral overexpression system coupled with scRNA-seq. The central challenge addressed is the absence of intra-pool negative controls in the publicly deposited barcode-cell mapping, which impedes recovery of validated TF-specific transcriptional signatures from the pooled single-cell perturbation screens. The authors developed a fully automated computational pipeline leveraging embryoid body (EB) cells as an external negative control, augmented by a background subtraction approach for artifact removal, to robustly delineate TF-specific regulatory programs despite incomplete experimental metadata.
Dataset Overview and Quality Control
The processed compendium comprises 254,519 cells from 30 experiments, including 8 pooled MORF screens and diverse single-TF overexpression samples. Rigorous QC yielded samples with favorable technical characteristics—median gene detection rates ranged from 2,500–4,200 per cell, and low mitochondrial content was consistently maintained. Unsupervised UMAP projections reveal clear segregation of experimental conditions and strong replicate concordance among the pooled screens.

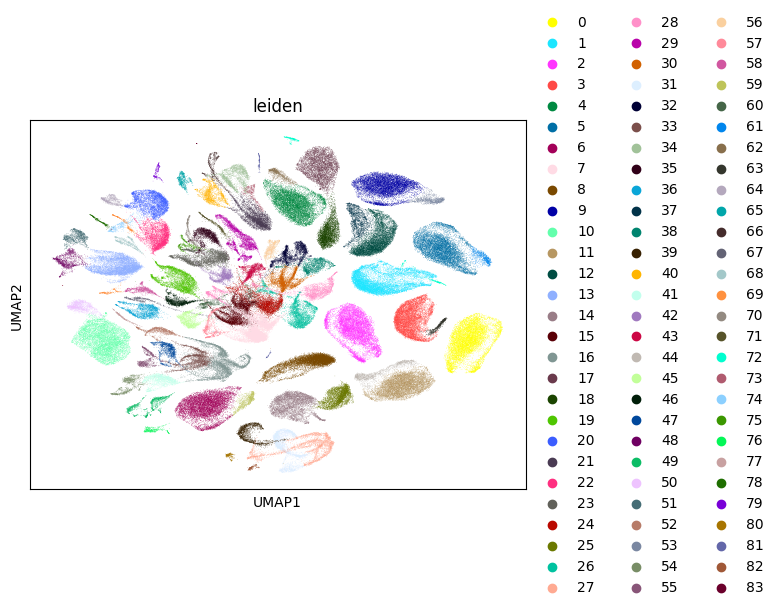
Figure 1: UMAP visualization colored by sample identity demonstrates separation of experimental conditions and tight overlap of pooled replicates.
Leiden clustering identified 83 transcriptionally discrete populations. This level of technical reproducibility is essential for downstream differential analyses.
Barcode Demultiplexing and TF Assignment
The demultiplexing procedure, based on the MORF barcode-to-TF assignment map, achieved an assignment rate of 79.2% for the pooled screens, corresponding to 60,997 cells mapped unambiguously to 87 TFs. Assignment rates were consistent across all replicates. The most abundant TFs by representation included HES5, ID4, HES1, and MEIS1.
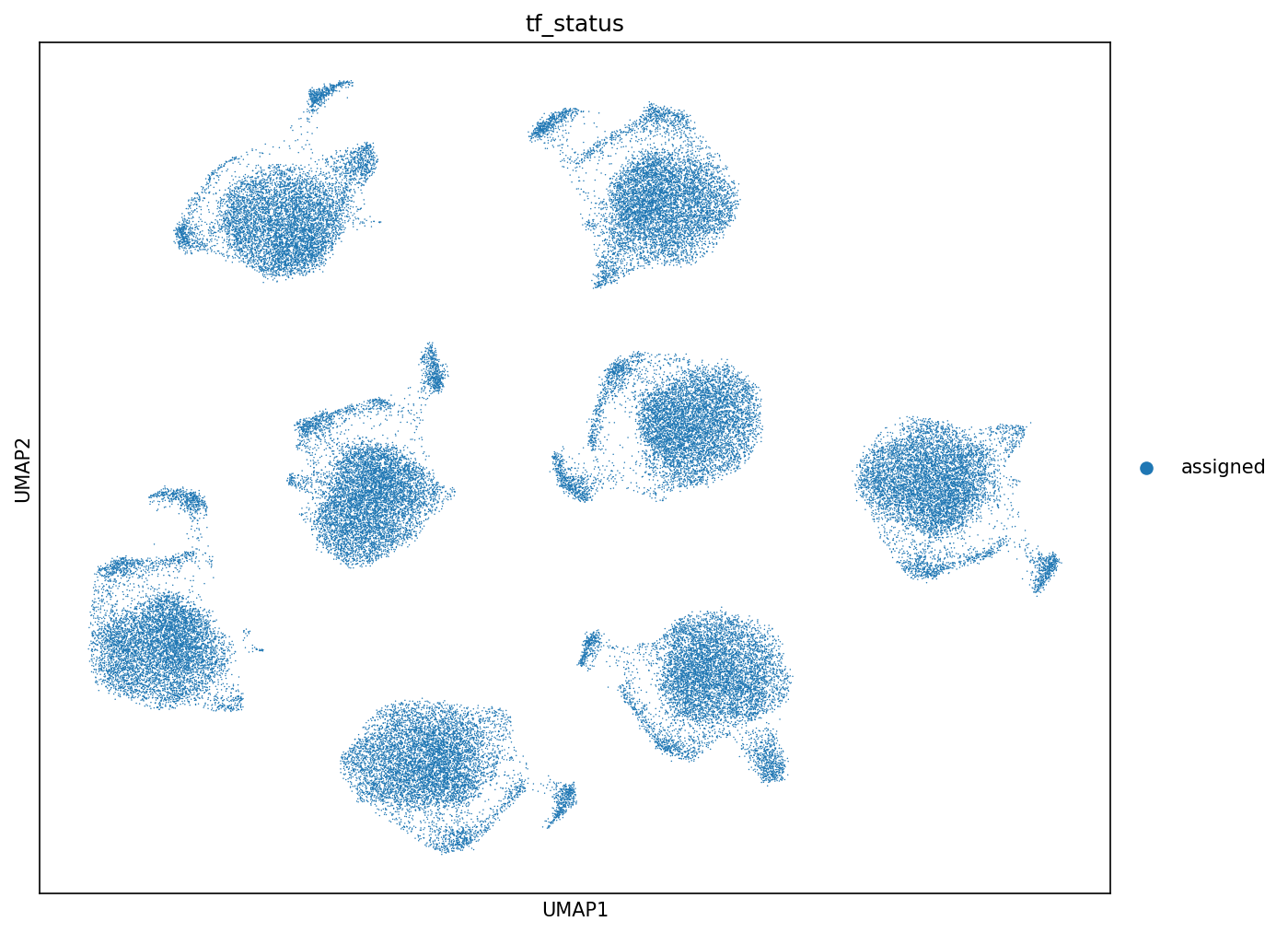
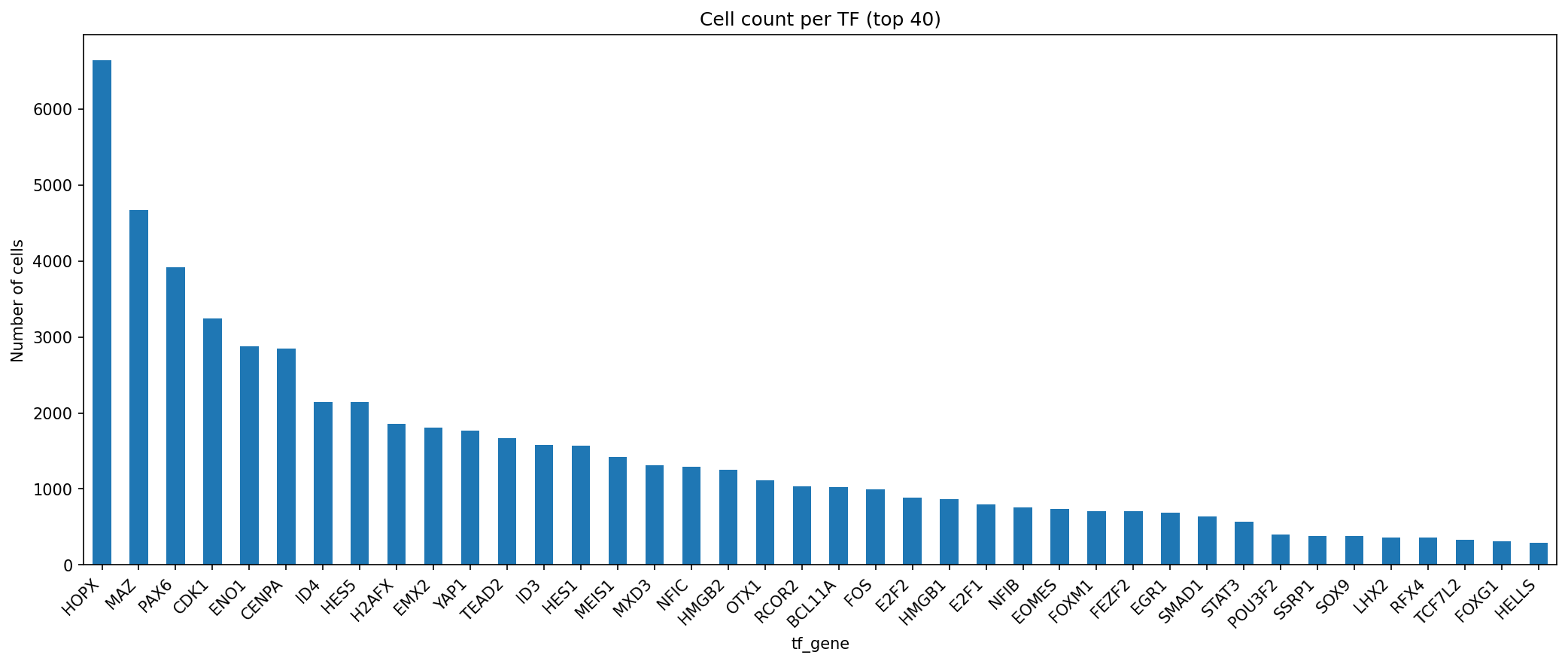
Figure 2: UMAP colored by demultiplexing status highlights the fraction of assigned, ambiguous, and undetected barcodes.
Ambiguous and unassigned cells were systematically excluded from all TF-specific analyses, eliminating confounding from undetermined perturbations.
Differential Expression: External Control and Artifact Subtraction
To counteract the missing in-pool negative controls (GFP/mCherry), the authors designated EB cells as the baseline. However, direct inter-batch comparisons conflate TF-specific transcriptional shifts with batch and transduction artifacts. The pipeline, therefore, introduces a crucial background subtraction step: genes identified as DE in ≥70% of TFs (1,946 genes) are classified as background artifacts and omitted from TF-specific DEG lists.
Applying this strategy, 59 of 61 TFs with sufficient representation (≥20 cells) displayed significant TF-specific differential gene expression (∣log2FC∣ > 0.5, FDR < 0.05), a twofold increase compared to the naive one-vs-rest approach, which detected only 27. The approach rescued substantial TF-specific signal that would otherwise be undetectable due to technical confounds.
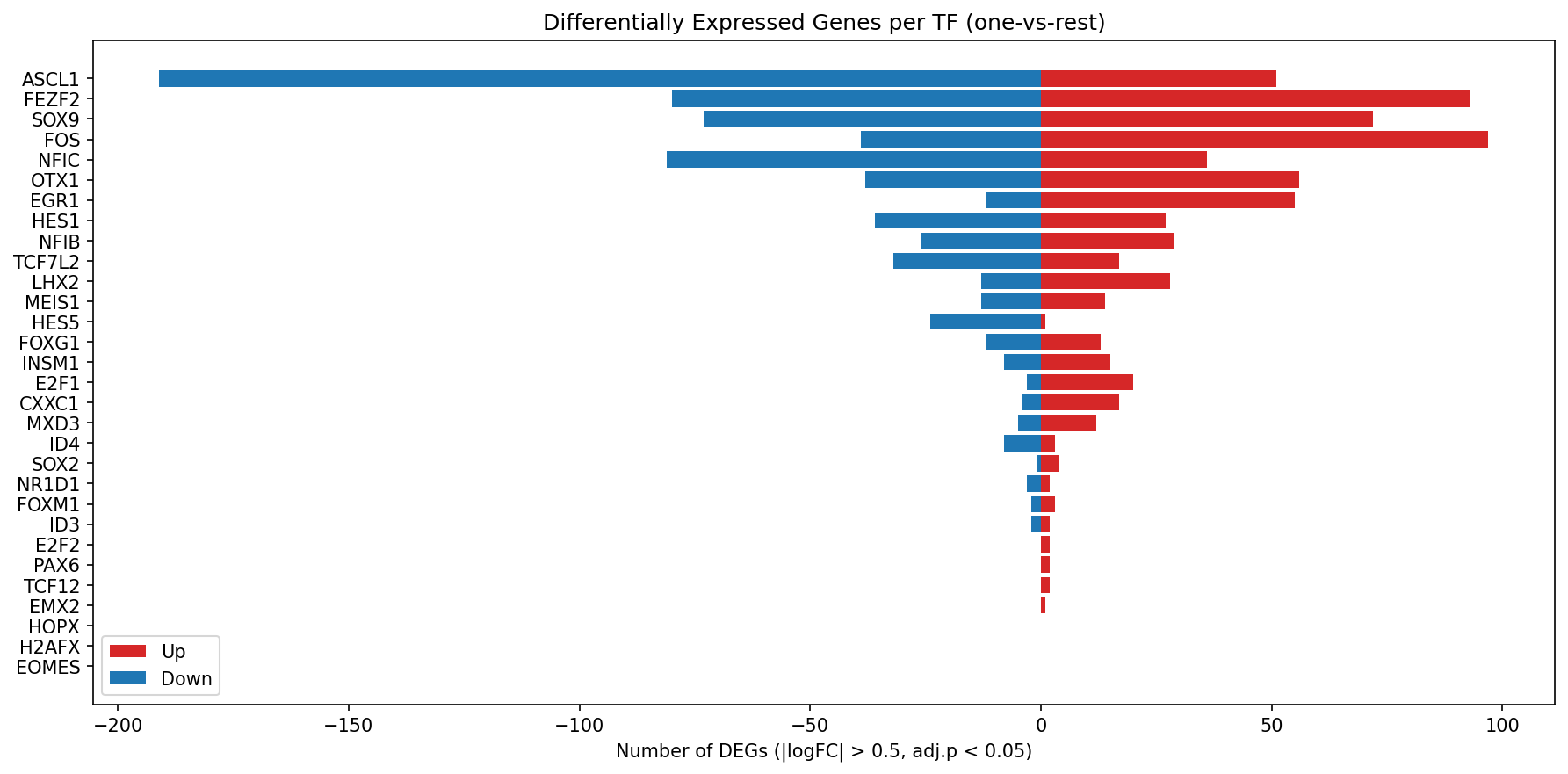
Figure 3: TF-specific DEGs per TF (vs. EB with background subtraction) reveal both the magnitude and directionality of regulatory activity for the top 30 TFs.
HOPX, MAZ, PAX6, FOS, and FEZF2 manifested as the strongest transcriptional remodelers, while TFs such as ASCL1 sharply dropped in apparent effect following artifact removal, highlighting the necessity of rigorous confound correction.
Pathway Enrichment and Biological Insights
ORA of condition-level DEGs revealed recurrent engagement of canonical pathways, including Wnt signaling, neurogenesis, axon guidance, EMT, and Hippo/YAP signaling—highlighting both shared and TF-specific regulatory axes within the differentiation context of the screens.
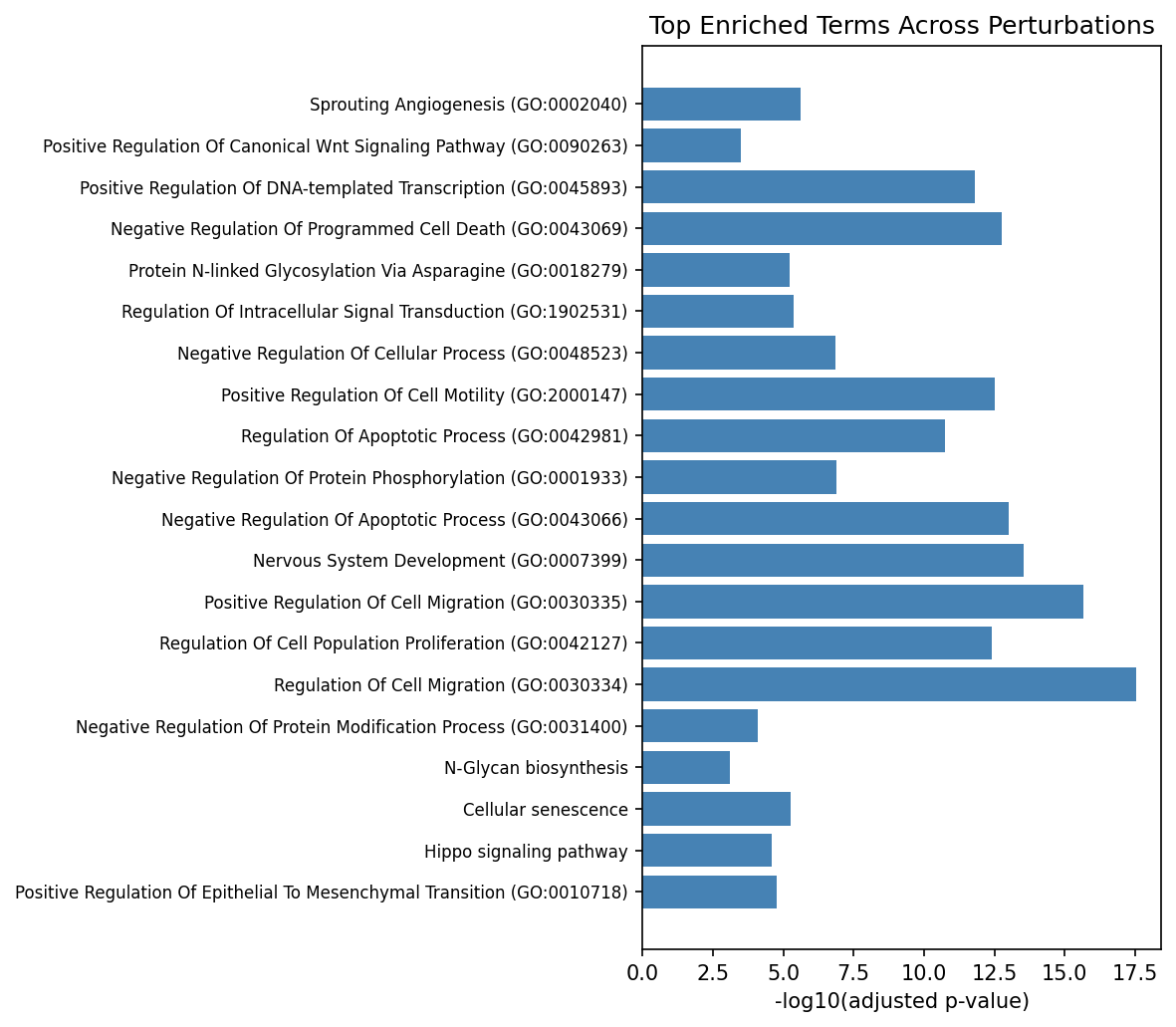
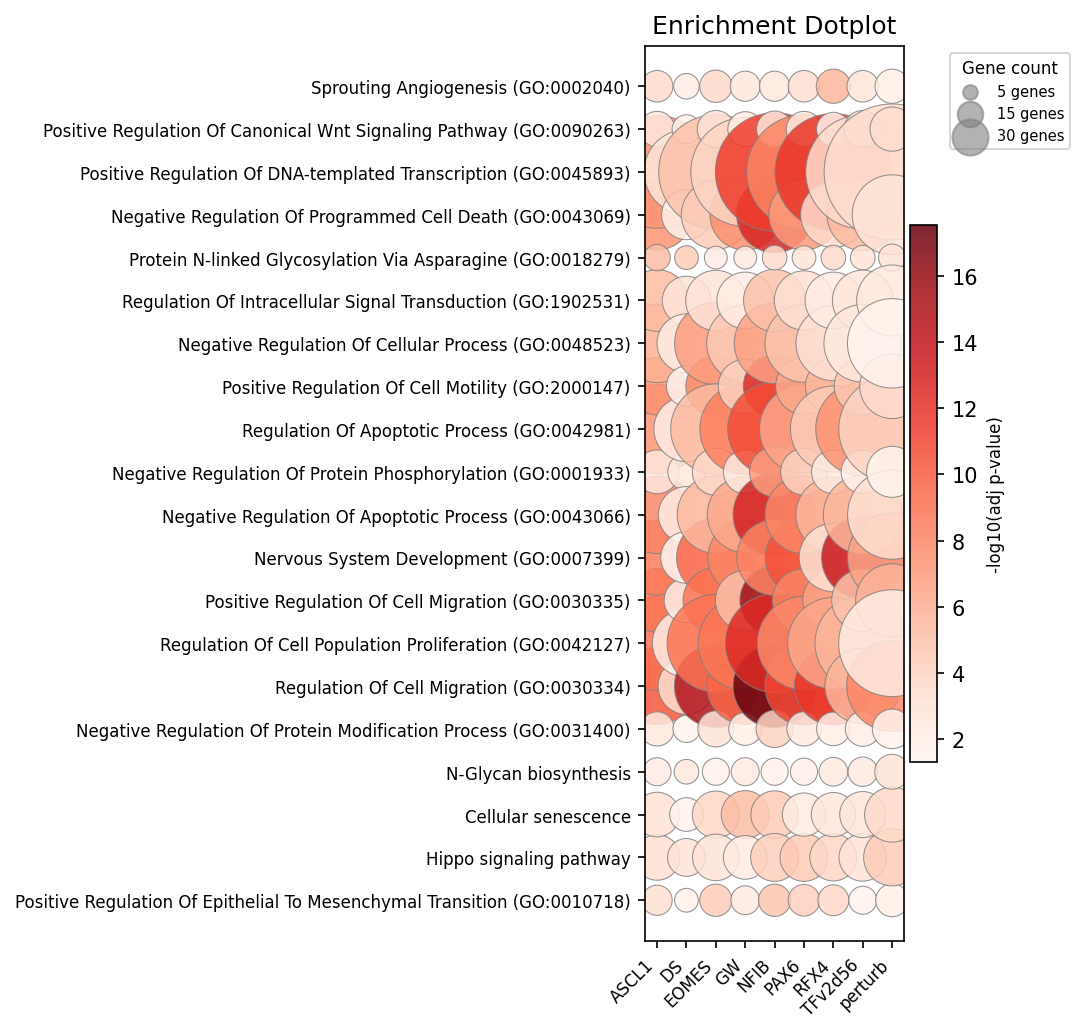
Figure 4: Top enriched GO/KEGG terms consistently recurrent across independent perturbation contexts.
Per-TF pathway analysis in the pooled context yielded significant enrichment for 13/61 TFs, with 207 pathways encompassing both established and previously underappreciated biological functions:
- FEZF2: Negative regulation of differentiation and neuron specification, reaffirming its role in cortical development.
- EGR1: Cardiac muscle and Hippo pathway specification, consistent with immediate-early TF roles in mechanotransduction.
- FOS: Focal adhesion and ECM remodeling.
- NFIC: Collagen biosynthetic regulation.
A systematic TF-pathway enrichment map delineates distinct modules, including metal ion response, muscle/cardiac, and adhesion/EMT clusters.
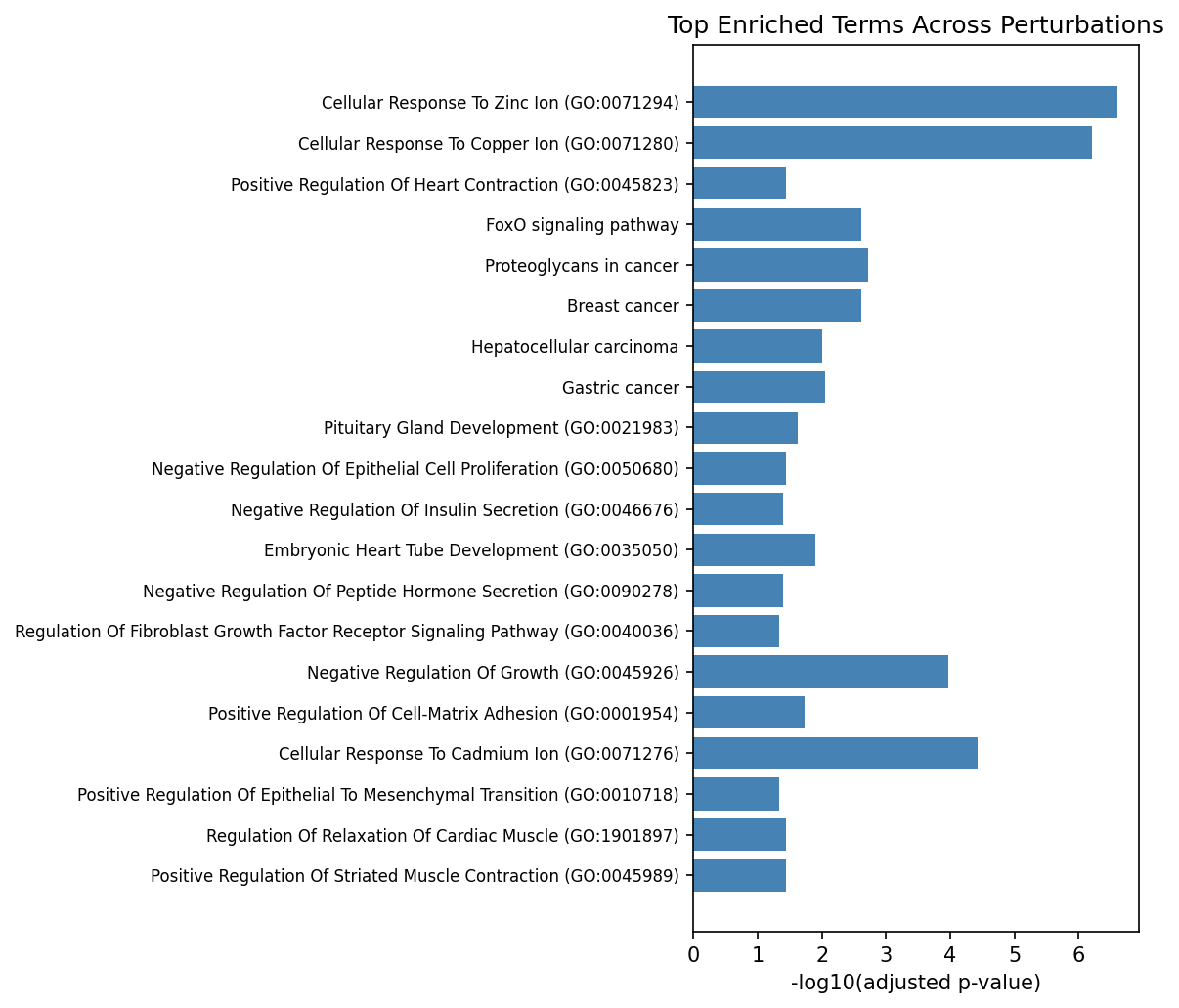
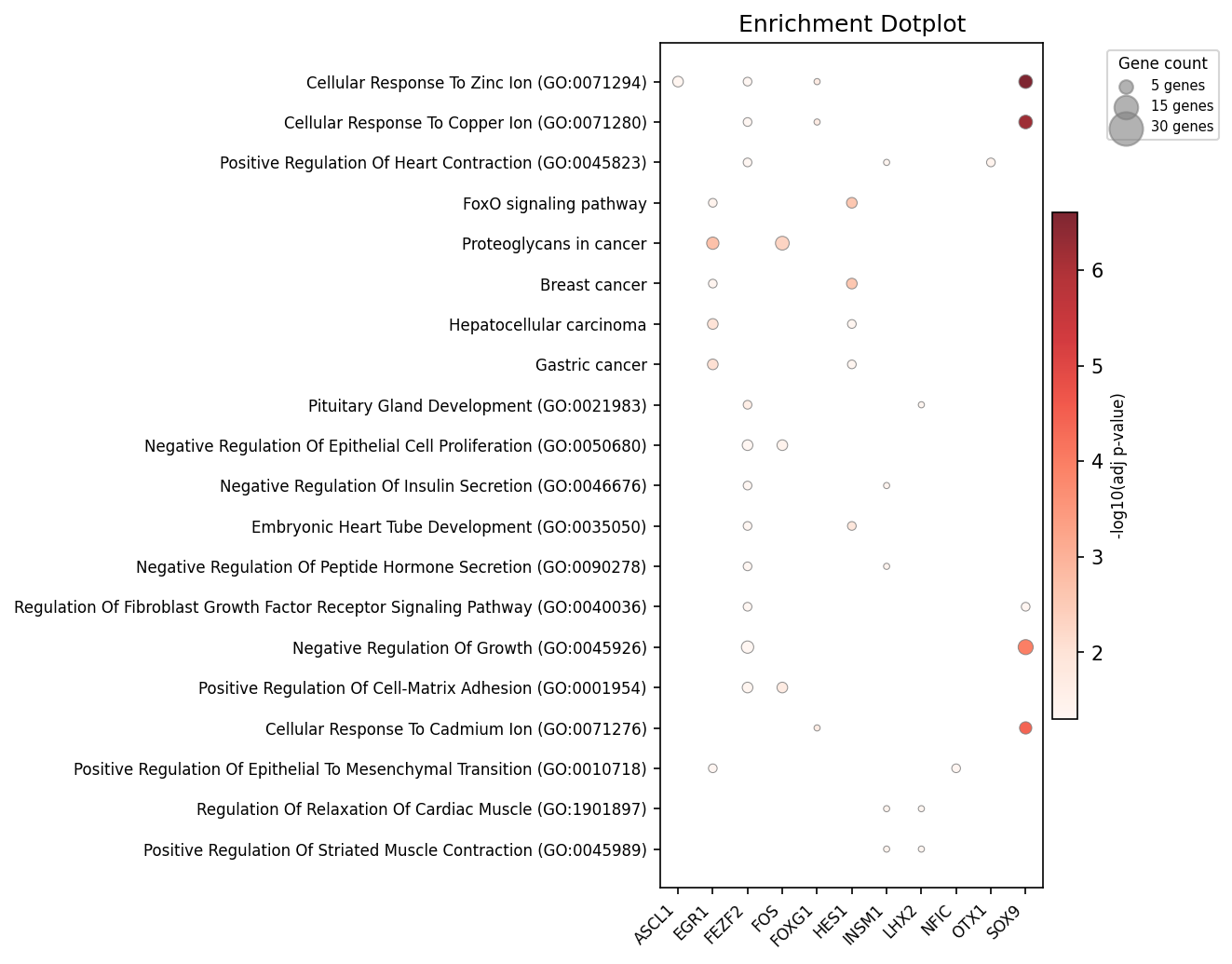
Figure 5: Top enriched terms across TFs, emphasizing convergent and divergent regulatory programs.
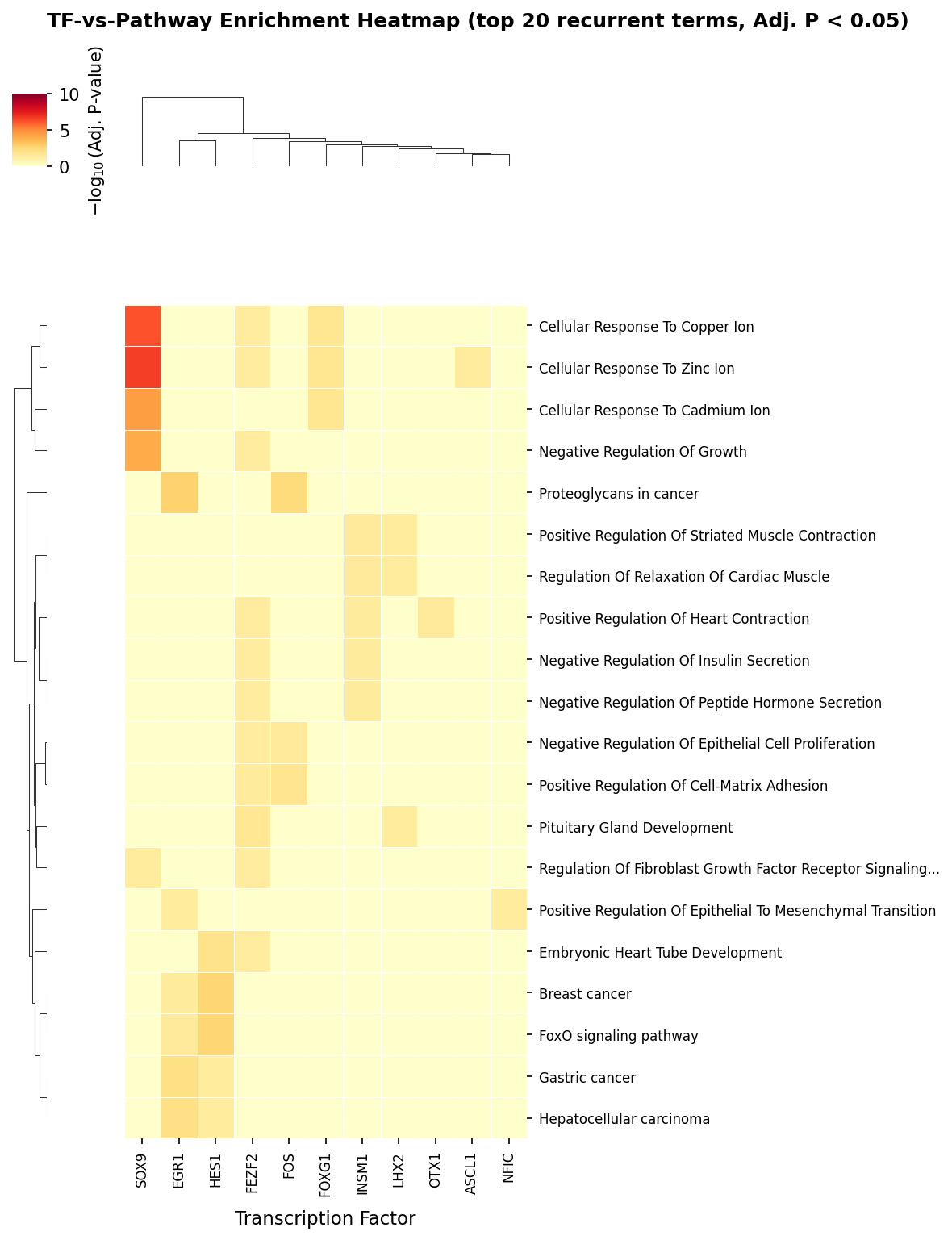
Figure 6: Clustered heatmap of TF-pathway enrichment shows functional modularity and shared effector circuits.
Assessment and Mitigation of Batch Effects
Batch integration using Harmony confirmed minimal inter-replicate batch effects, as UMAP topologies before and after correction exhibited negligible shifts. This supports the validity of direct comparisons using aggregate EB controls.
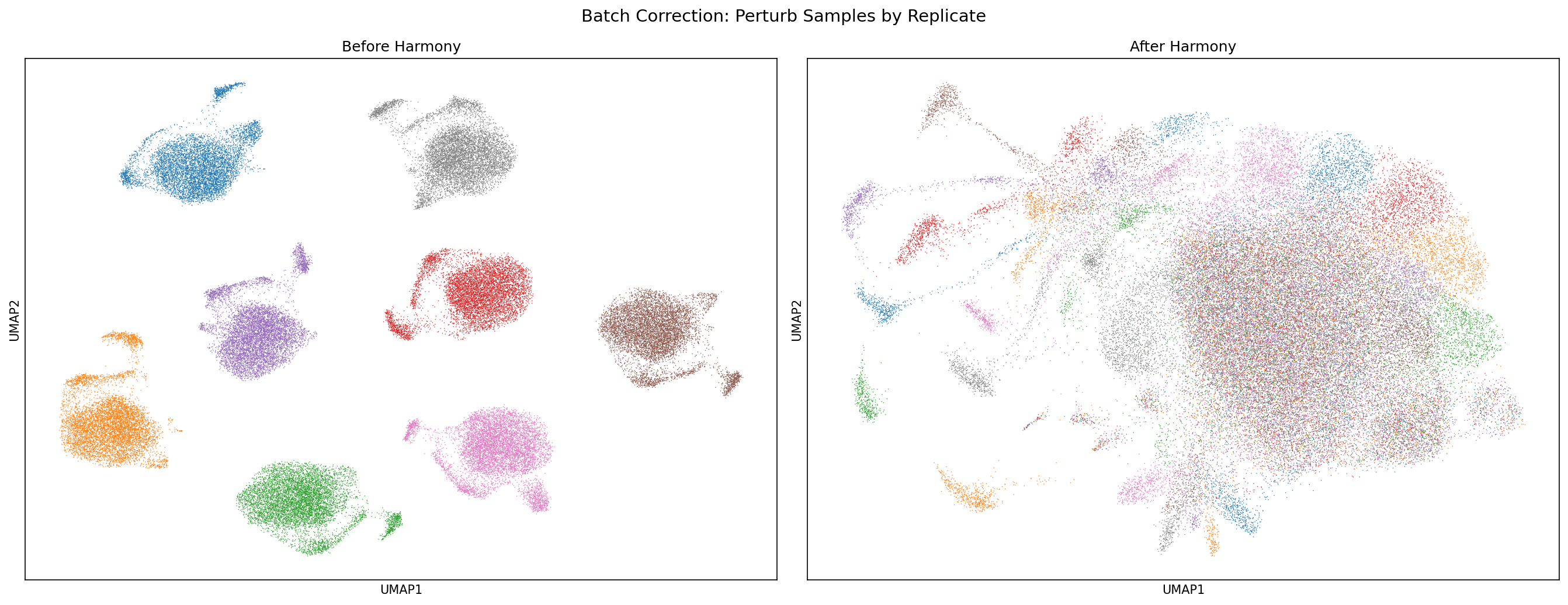
Figure 7: UMAPs before and after Harmony-based batch correction demonstrate negligible batch effect, validating analytic integrity across screen replicates.
Validation Against Published Rankings
The recovered TF effects were quantitatively benchmarked against the original TF ranking by Joung et al. A significant negative correlation (Spearman ρ=−0.316, p=0.013) between the number of background-subtracted DEGs per TF and the published scRNA-seq rank substantiates the fidelity of the re-analysis and the specificity of detected signatures.
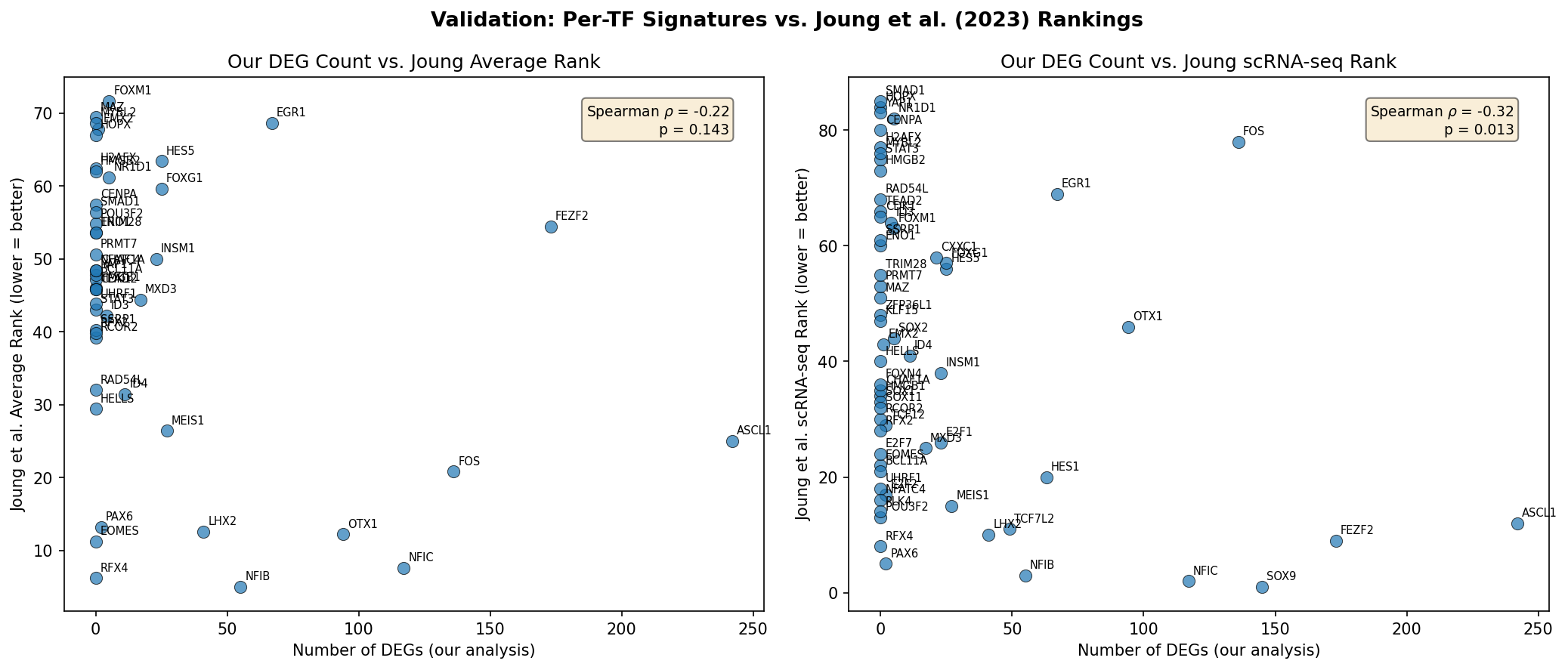
Figure 8: Validation of per-TF signatures against published rankings, denoting robust recapitulation of prior characterization despite missing controls.
Implications and Future Directions
This analysis underscores that robust TF-specific regulatory programs can be extracted from pooled perturbation atlases even with incomplete in-pool controls, provided external controls and principled artifact modeling are applied. The presented pipeline is entirely reproducible, facilitating broader adoption and extension. The results enable more granular biological hypotheses regarding TF-mediated reprogramming, particularly for previously uncharacterized factors.
Practical implications include application in re-analyses of other public perturbation datasets with missing metadata, the establishment of methodological standards for artifact subtraction, and the possibility of integrating advanced effect estimation frameworks (e.g., mixscape, CINEMA-OT) to further refine signal disambiguation.
Conclusion
The authors demonstrate that TF-specific transcriptional signatures can be robustly recovered from the deposited Human TF Atlas via an automated, artifact-corrected pipeline employing external EB controls and stringent background subtraction. This doubles the number of detectable TF effects over naive strategies and maintains significant concordance with published benchmarks. The work provides both an analytic framework and a resource for rescuing interpretable biology from public pooled perturbation datasets, even in the presence of incomplete experimental annotation.