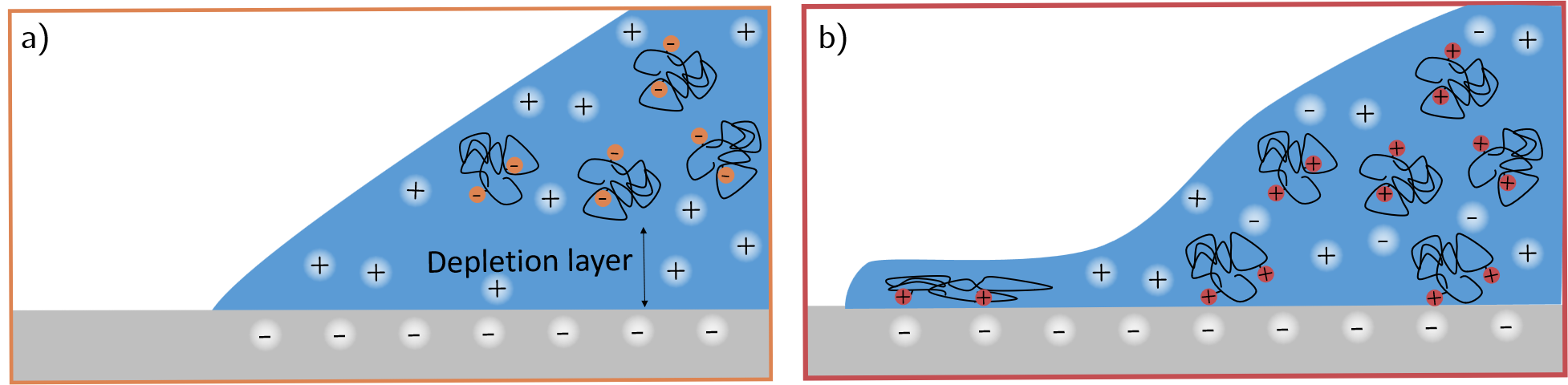
- The paper demonstrates that polymer charge governs the transition between interfacial depletion (anionic) and adsorption (cationic/non-ionic), driving receding contact line instability.
- High-speed reflection microscopy and quantitative rheometry reveal that cationic and non-ionic drops exhibit larger deformations, increased contact angle hysteresis, and filament formation compared to anionic drops.
- The results suggest that tuning polymer charge and pH can effectively control drop friction and patterned deposition for applications in printing and surface engineering.
Introduction
Controlling the wetting dynamics and instabilities of non-Newtonian drops on solid substrates is pivotal for a variety of technological processes involving printing, coating, and biofluids. While the macroscopic behavior of sliding viscoelastic drops—such as reduced translational velocities and enhanced elongation—is well-documented, the microscopic origins underpinning contact line instability remain unresolved. This work bridges that gap, leveraging high-speed, high-resolution reflection microscopy to directly observe the receding contact line morphology of polyacrylamide-based polyelectrolyte drops as they slide on hydrophobic Teflon AF and PDMS surfaces. The focus is on elucidating the interplay of rheological parameters and interfacial polymer/surface interactions, particularly the role of polymer charge in the nucleation and evolution of receding contact line instabilities leading to filament formation.

Figure 1: Experimental setup for simultaneous side- and bottom-view imaging, enabling direct visualization of contact line deformations in sliding drops.
Experimental Approach
Drops of three polymer types—non-ionic polyacrylamide, anionic polyacrylamide-co-acrylic acid, and cationic polyacrylamide-co-chloro-methylated acrylamide—were studied at a fixed concentration in water. Hydrophobic glass slides (Teflon AF or PDMS coated) were inclined at adjustable angles between 20° and 45°, inducing drop motion under gravity. An inverted epifluorescence microscope in reflection mode, synchronized with a side-view camera, allowed for in situ quantification of advancing/receding contact angles, contact angle hysteresis, and filament dynamics at the receding rim.
For all polymeric systems, the advancing contact line remained macroscopically smooth and was essentially indistinguishable from that of pure water. However, contact line instability was prominent at the receding edge, with the morphology and magnitude of deformation dependent on polymer charge:
Quantitative Analysis of Instability
Velocity analysis, vortexing rheometry, and scaling using effective capillary number, Ca=σηeff(γ˙)U, showed substantial differences in drop mobility: anionic drops moved faster, maintaining capillary numbers an order of magnitude larger than the other two. After normalizing by viscosity, non-ionic and cationic drops were substantially slowed, indicating an elevated friction force at the receding edge.
Contact angle hysteresis, extracted as the difference between advancing and receding angles, was sharply increased for non-ionic and cationic systems, exceeding 70° at high tilt, compared to the stable 30–40° observed for anionic drops. This effect was primarily traced to a reduction in the receding contact angle, supporting a mechanism of increased friction at the contact line for non-ionic and cationic drops.
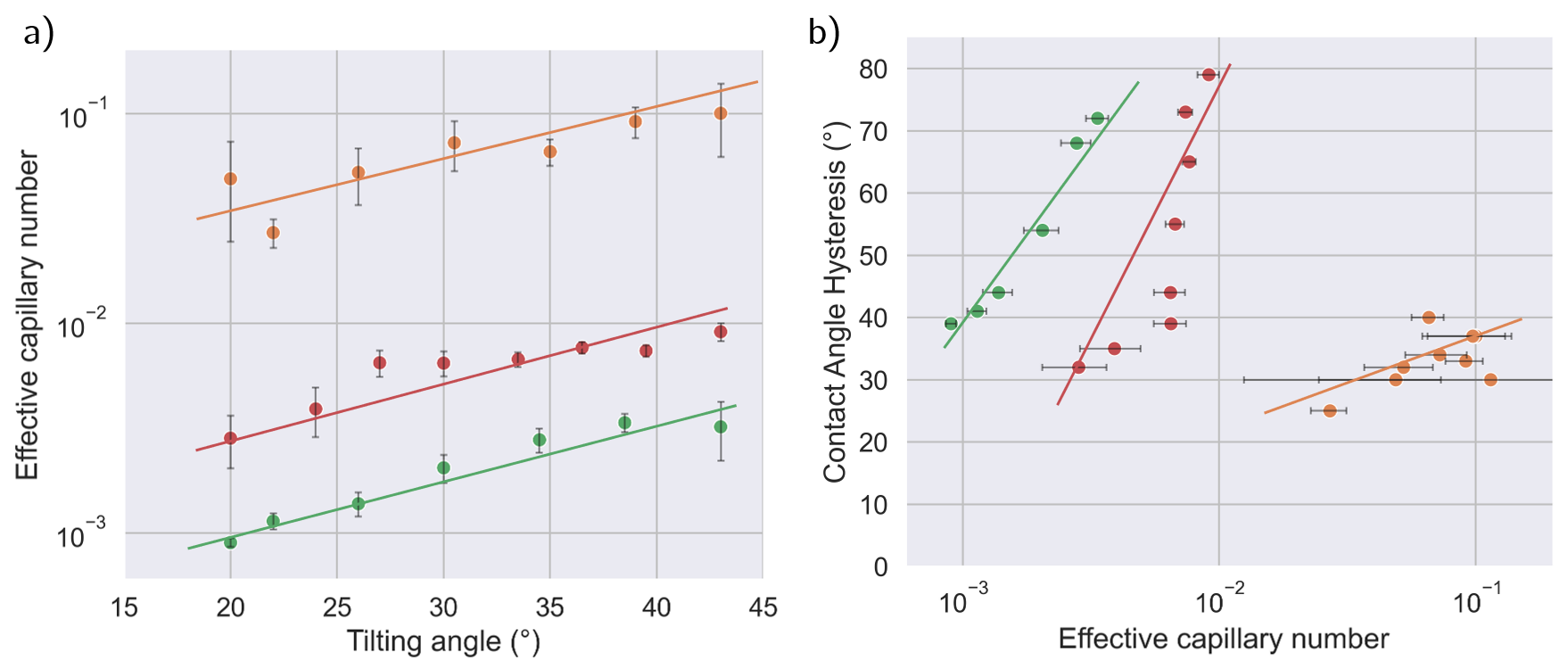
Figure 3: (a) Effective capillary number as a function of tilting angle. (b) Contact angle hysteresis rises with increasing capillary number for non-ionic and cationic systems.
Systematic mapping of filament length (L) and wavelength (λ) revealed both to grow with increasing tilt, plateauing at L≈175μm and λ≈90μm for cationic drops. These spacings are consistent with prior studies of receding contact line instability in both Newtonian and viscoelastic thin films, where the dominant instability is a viscoelastic-modified Rayleigh-Plateau mechanism [sharma_newtonian_2025, deblais_taming_2016]. Notably, anionic polymers failed to produce extended filaments regardless of the velocity or tilt, emphasizing the decisive role of charge-mediated surface interactions.
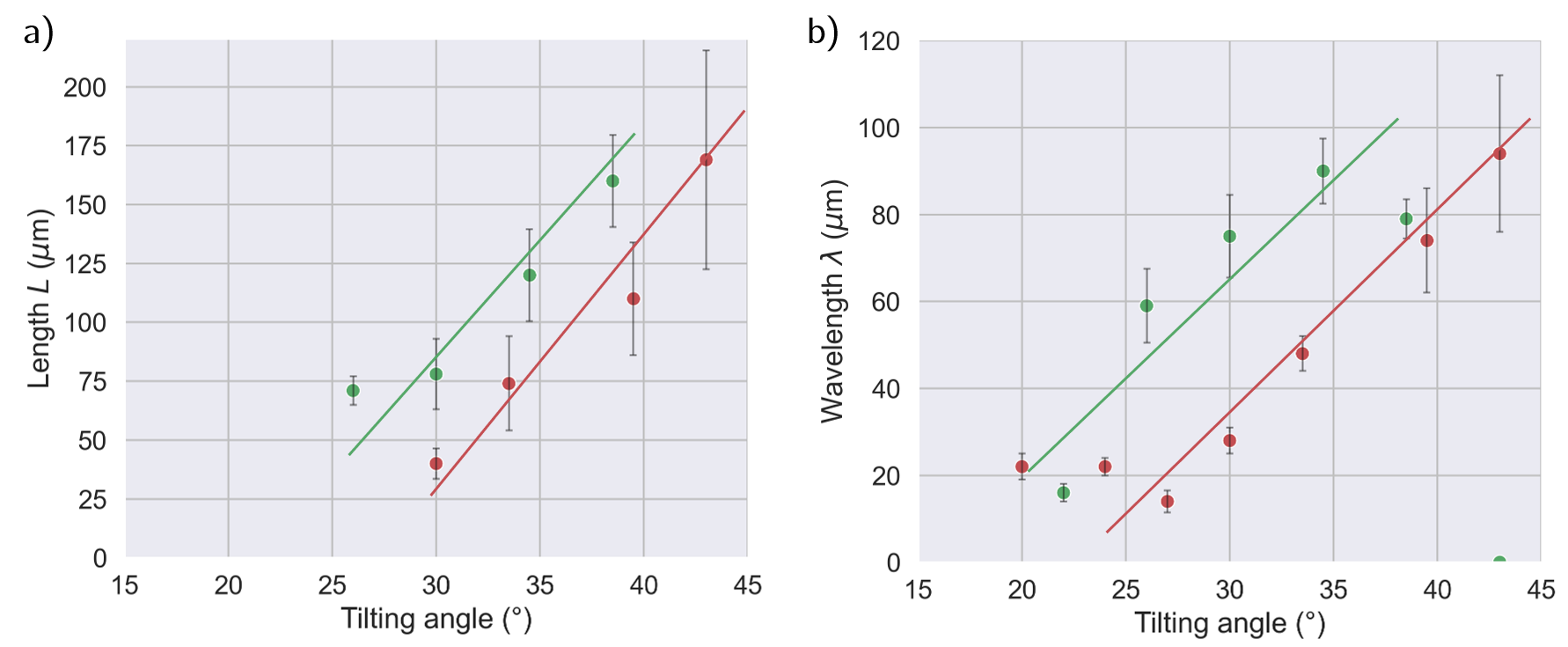
Figure 4: Filament length and spacing (L, λ) as a function of tilt angle for non-ionic and cationic polymers.
Mechanistic Insights: Rheology vs. Interfacial Adsorption
Viscoelastic hydrodynamics alone cannot account for the observed differences: the anionic system had the highest relaxation time but the weakest instability, and theoretical treatments predict that viscoelasticity primarily affects the receding contact angle while leaving the advancing rim unperturbed [kansal_viscoelastic_2024, bartolo_dynamics_2007].
Instead, direct evidence from bottom- and side-view microscopy, coupled with Scanning Electron Microscopy, points to interfacial polymer adsorption as the controlling factor:
Influence of pH and Substrate Chemistry
Tuning the pH of the non-ionic polyacrylamide solution showed a direct correlation between protonation, interfacial adsorption, and filament formation. Lower L0 (stronger adsorption, partial cationization) promoted both tail elongation and filament nucleation; at high L1, behavior mirrored that of the anionic system, with minimal deposition or instability.
The results were qualitatively robust on PDMS-coated slides—another hydrophobic but more compliant coating—though kinetic friction was generally increased and cationic drops tended to pin even at high inclinations. Nevertheless, the charge-dependence of instability was recapitulated, underlining the centrality of electrostatic adsorption and depletion in this phenomenon.

Figure 6: pH-tunable adsorption effects for non-ionic polyacrylamide; filaments and tails are prominent at pH 5 but suppressed at pH 11.
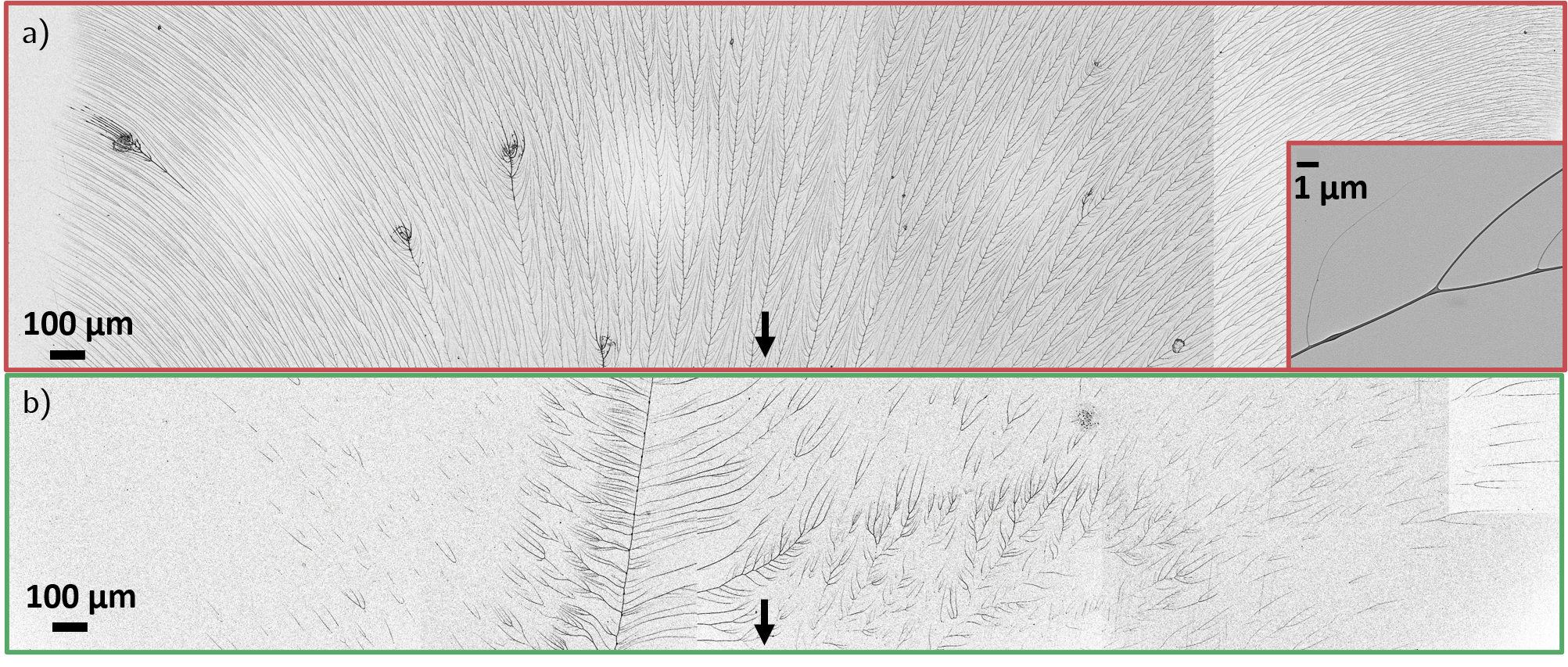
Figure 7: SEM visualizations of cationic (left) and non-ionic (right) polymer deposition after a single drop passes; filamentary patterns are evident for cationic systems.
Polymer Deposition and Extensional Rheology
SEM imaging post-drop revealed that cationic polymer filaments dry to form robust, micron-scale deposits at the rear of the drop, aligned at angles up to 45° relative to the drop path. Non-ionic polymers produce sparser, tail-based deposits, while anionic systems leave virtually no residue detectable at this scale.
Analysis of filament thinning shows an initial extensional viscosity well above the critical value for filament survival (estimated as L2), with viscoelastic stabilization permitting elongated filaments prior to breakup by Rayleigh-Plateau-type instability. Thinning and break-up are thus temporally delayed, resulting in microdroplet arrays bound by residual polymer filaments.
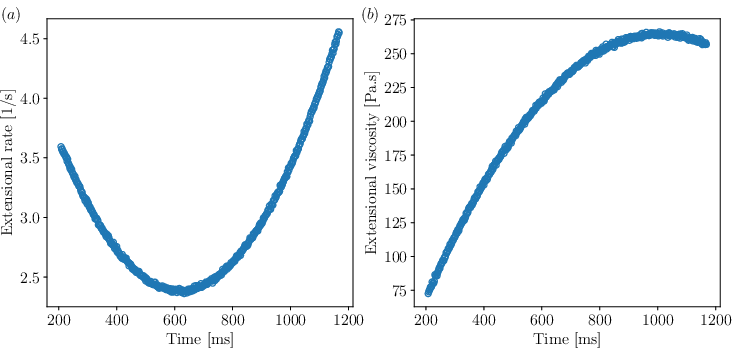
Figure 8: Extensional rate (a) and extensional viscosity (b) display the viscoelastic character of the anionic polymer solution.
Theoretical and Practical Implications
These results validate and extend recent theoretical predictions that receding film instability and filamentation require average film thicknesses below a threshold governed by van der Waals and capillary forces, but that the macroscopic manifestation—filament survival, length, and interspacing—is determined by the balance of hydrodynamic and surface forces, critically by polymer adsorption at the solid–liquid interface [sharma_newtonian_2025, de_gennes_polymer_1981, guyard_near-surface_2021].
Key findings are:
- Polymer charge controls the transition between depletion (anionic, low instability) and adsorption (cationic/non-ionic, high instability or filamentation).
- Contact angle hysteresis and kinetic friction are strongly modulated by interfacially adsorbed polymers, with extensions into the design of polymeric coatings, printing mechanisms, and surface patterning strategies.
- The receding contact line stability is a quantifiable indicator of interfacial polymer phenomena and can be rationally tuned with solution chemistry and substrate modification.
Conclusion
This work establishes that, for sliding viscoelastic drops of polyacrylamide-based polymers, receding contact line instability—and thus filament, microdroplet, and surface deposit formation—are governed not simply by bulk viscoelasticity, but by charge-mediated adsorption at the solid–liquid interface. The interplay between the sign of the polyelectrolyte, substrate charge, and L3-induced protonation defines a continuum between depletion and adsorption regimes, directly controlling instability nucleation and surface patterning. This reveals new avenues for rational control of non-Newtonian drop dynamics and functional films with relevance to advanced manufacturing, microfluidic design, and surface engineering.