- The paper introduces a bond-centered DFT+DMFT framework that unifies the description of VO2’s various structural and electronic phases.
- It demonstrates how electronic correlations and lattice distortions drive simultaneous metal-insulator transitions in both Peierls and Mott regimes.
- The study highlights the critical role of lattice strain and bonding effects in stabilizing M2 and other transitional phases, paving the way for tailored device applications.
Unified DFT+DMFT Description of VO2: Bond-Centered Orbital Approach
Introduction
The MIT in vanadium dioxide (VO2) has been a canonical platform for exploring the interplay between electronic correlations and structural instabilities. Above TMIT≈340 K, the rutile (R) phase is metallic, while cooling triggers a transition to the monoclinic M1 phase, which is insulating and features vanadium dimerization and zigzag distortions. Less studied, but equally significant, are the structurally distinct M2 and T phases, which challenge the prevalent dichotomy between Peierls and Mott-Hubbard mechanisms. This paper presents a unified first-principles framework—employing DFT+DMFT with unconventional bond-centered correlated subspaces—that consistently captures the electronic properties and MIT across the full structural phase diagram of VO2, obviating the need for prepatterning into dimerized/undimerized chains.
The central technical advancement is the adoption of a bond-centered Wannier orbital basis, constructed from a1g and egπ derived functions hybridized between adjacent vanadium ions along the c axis. This symmetry-agnostic basis enables seamless treatment of Peierls-dimerized, zigzag-distorted, or mixed vanadium chains across all VO2 phases without explicit cluster partitioning. The full structural configurational space is parameterized by collective distortions η1 and η2 within (110)-type planes, tracing pathways between R, M1, M2, and T phases.
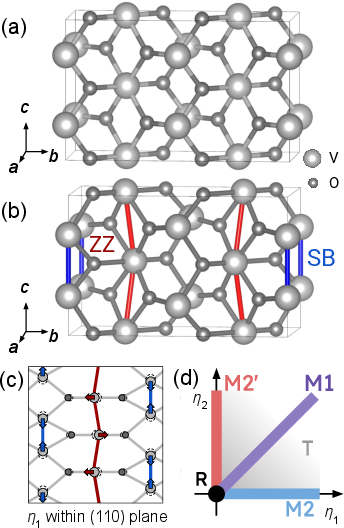
Figure 1: Panel (a) and (b): R and M2 unit cells depicting short-bond (SB) pairs, zigzag (ZZ) chains; panel (c): 20 distortion mode; panel (d): schematic 21 phase diagram indicating R, M1, M2, and T phases.
DFT+DMFT calculations are implemented using the Quantum ESPRESSO, Wannier90, and TRIQS/solid_dmft stack, with the DMFT impurity problems formulated via the Hubbard-Kanamori Hamiltonian on each bond-center. Interaction values are set according to cRPA estimates to maintain realism (22 eV, 23 eV), but in-depth explorations are conducted up to 24 eV to probe both metallic and insulating regimes.
Electronic Structure of the M2 Phase
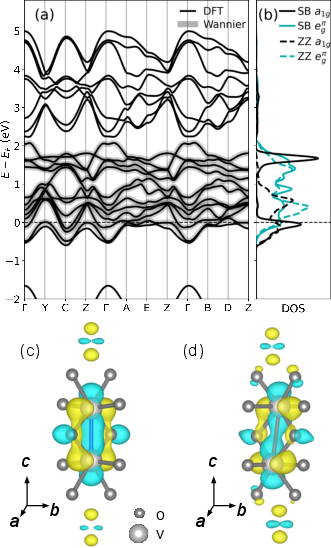
Figure 2: (a) DFT and Wannier band structures for M2; (b) orbital-projected DOS for M2 SB and ZZ sites.
In the M2 phase, the vanadium chains partition into two electronically distinct types: Peierls dimerized (SB/LB) and zigzag-distorted (ZZ). DFT+DMFT calculations in the bond-centered basis yield a correlated insulating solution wherein SB chains host singlet-paired 25 electrons (as in the M1 phase), while the ZZ chains manifest a Mott insulating state with local magnetic moments. Notably, these different insulating motifs are strongly coupled: the MIT occurs concomitantly for both chain types as a function of 26 or structural distortion with no site-selective intermediate regime.
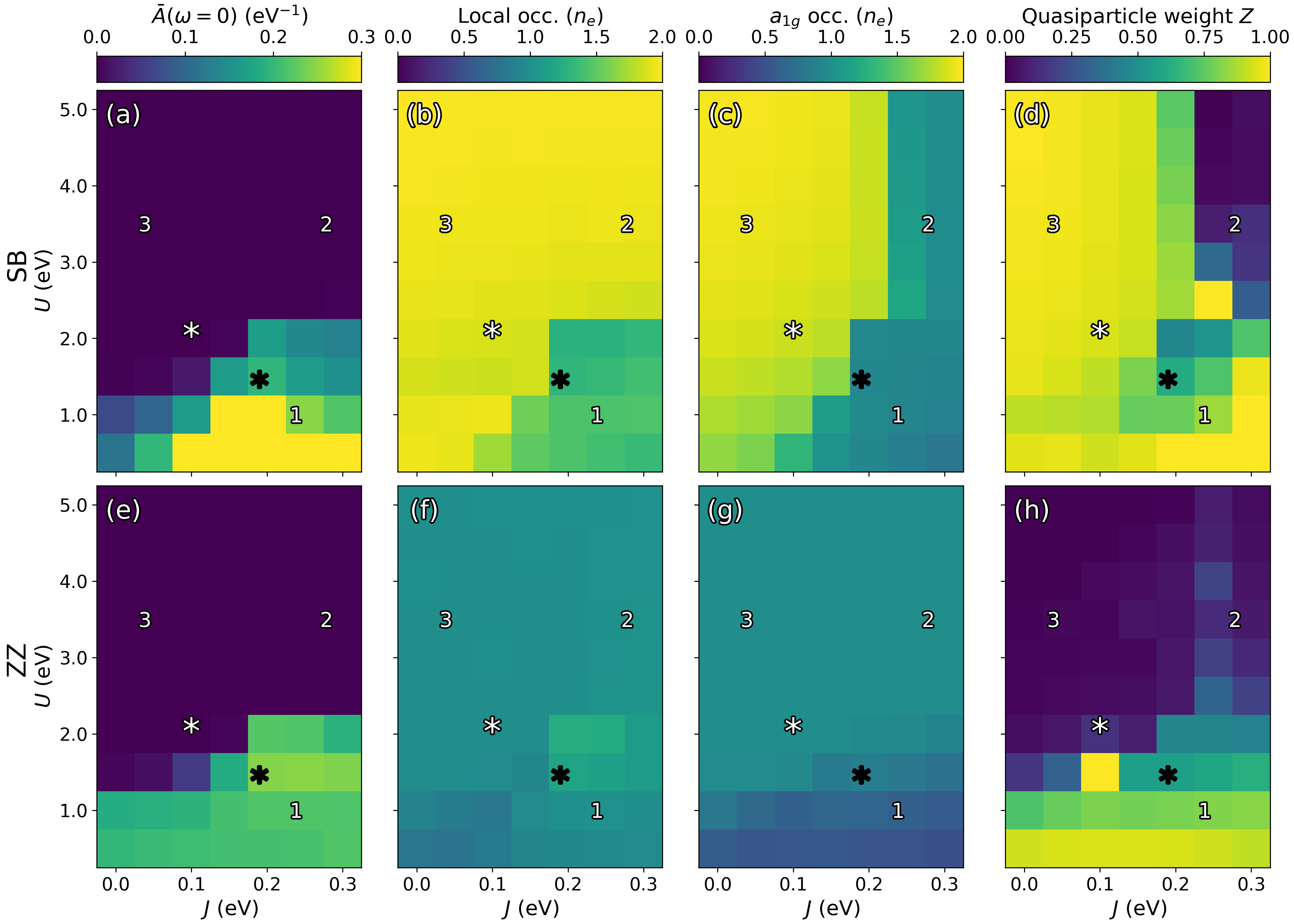
Figure 3: Local observables versus 27 and 28 for SB (a-d) and ZZ (e-h) sites; panels include zero-frequency spectral weight, total occupation, 29 occupation, and TMIT≈3400 quasiparticle weight TMIT≈3401.
The phase diagram reveals three regimes: (1) metallic at low TMIT≈3402, (2) a mixed insulator with Mott-triplet SB and Mott ZZ chains at higher TMIT≈3403, and (3) physical M2, where dimerized chains form weakly correlated singlets and ZZ chains are Mott insulating at moderate TMIT≈3404. The realistic cRPA parameter window places the physical system near the regime boundaries, indicating sensitivity to interaction strength, but the adopted TMIT≈3405 eV, TMIT≈3406 eV reliably stabilizes the experimentally consistent dual-insulator regime.
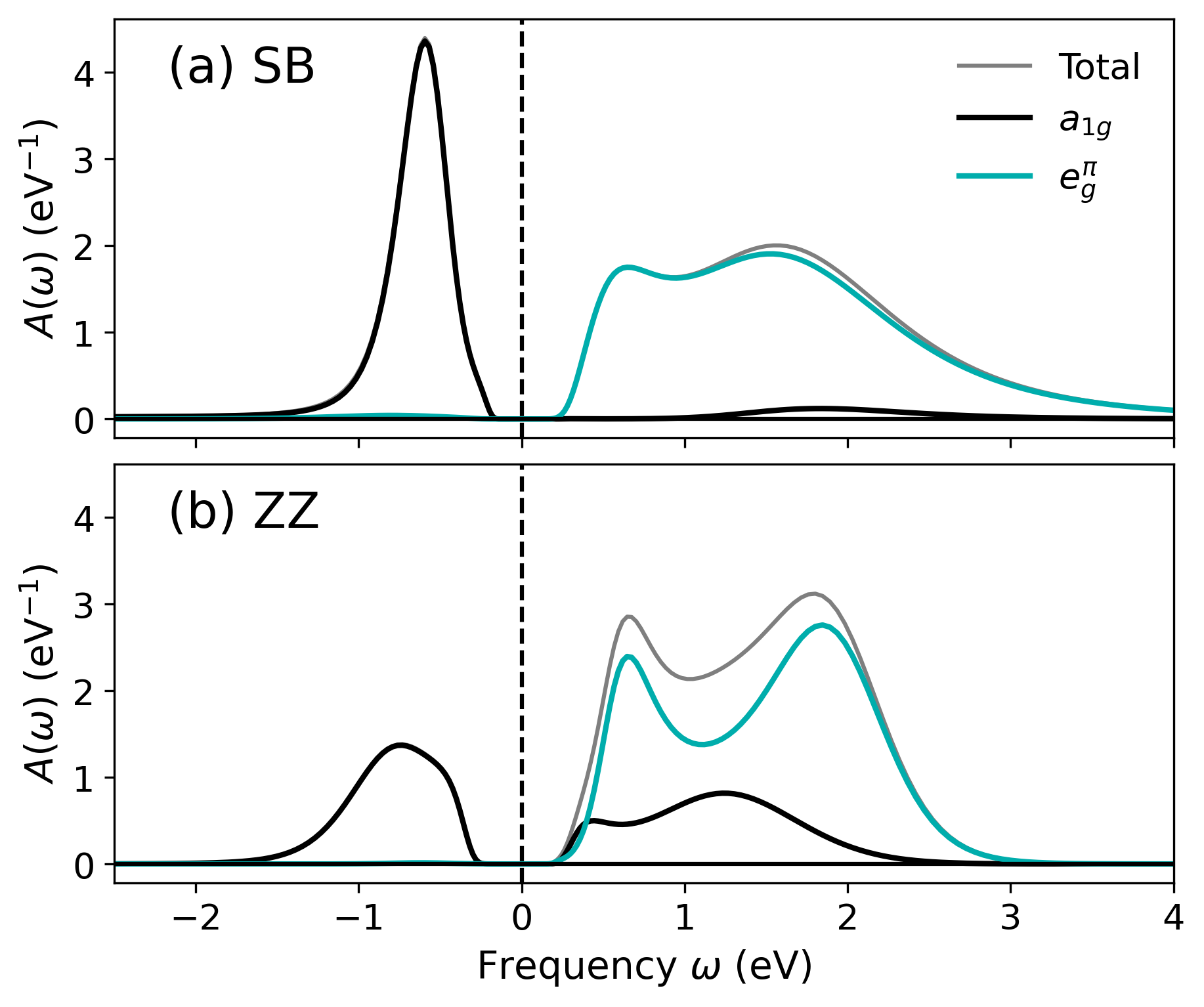
Figure 4: Local spectral functions for M2 SB and ZZ sites at TMIT≈3407 eV highlighting filled TMIT≈3408 in SB and Mott gap in ZZ.
Evolution Across Structural Distortions
Systematic interpolation between R, M2, and M1 phases using the bond-centered formalism elucidates the evolution of both electronic and energetic properties. Upon increasing the M2 distortion from R, metallicity persists up to a critical point, beyond which simultaneous localization occurs on both SB and ZZ chains, as reflected by the sharp reduction in zero-frequency spectral weight and integer TMIT≈3409 fillings (two and one electron(s) on SB and ZZ chains, respectively).
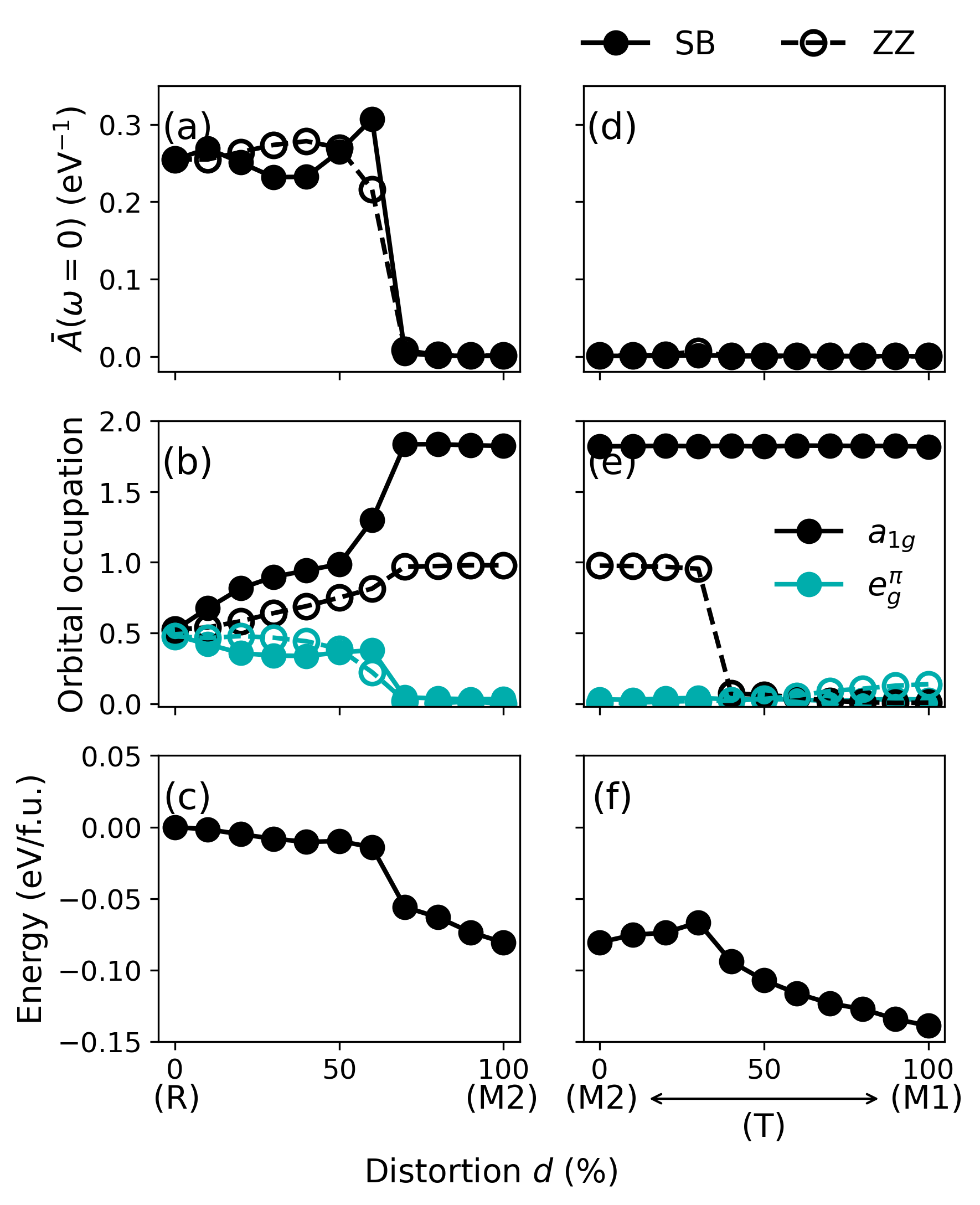
Figure 5: Evolution of spectral weight, orbital occupancy, and relative energy along R–M2 and M2–M1 (via T) structural paths for both SB and ZZ bonds.
The M2 phase emerges as a local energy minimum in the structural landscape, but globally remains less stable than M1. Along the M2–T–M1 distortion path, an abrupt transition occurs between distinct electronic ground states: from dual (SB/ZZ) insulators in M2/T to exclusively dimerized singlet insulator in M1. The theoretical observation of a sharp first-order transition is in line with experimental reports of M2–T transitions and continuous T–M1 crossovers.
Decoupling Unit Cell and Internal Distortion Effects
Separate analysis of lattice parameters and internal distortions reveals that monoclinic strain (20 angle) and 21 axis elongation both significantly reduce the critical 22 for insulating behavior and energetically favor the M2 phase. Conversely, imposing M2-type internal V positions within the R cell destabilizes the insulating state and local minimum.
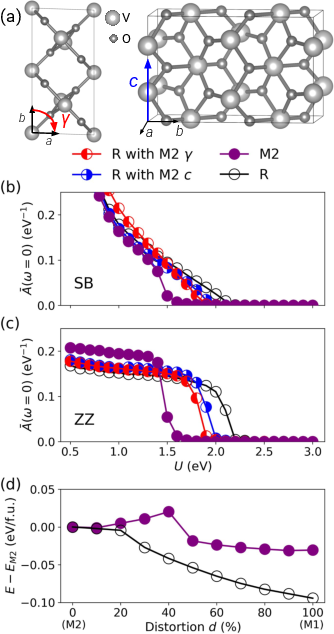
Figure 6: (a) M2 structure with highlighted strain; (b,c) spectral weights for various unit cell environments; (d) total energy versus internal distortion for R and M2 lattice settings.
This analysis underscores that unit cell deformations—often neglected or oversimplified—play a crucial role in driving and stabilizing the M2 insulator.
Isolated Dimerization vs. Zigzag Distortion
The study of hypothetical structures with exclusive dimerization (SB-only) or zigzag (ZZ-only) chains demonstrates that the former supports a Peierls-like MIT to a singlet insulator at relatively low 23 and modest distortions, while the latter requires significantly larger 24 to ignite a Mott transition, even at exaggerated zigzag amplitudes.
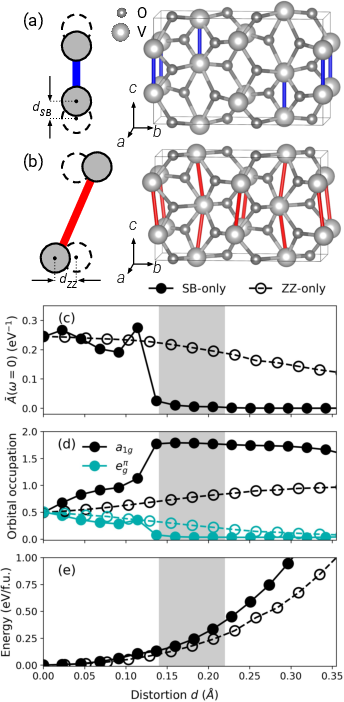
Figure 7: (a) SB-only, (b) ZZ-only structural models; (c-e) spectral weight, occupancy, and energy as a function of distortion for both models.
However, in the real M2 phase, the two transition channels are coupled: the MIT always occurs simultaneously for both SB and ZZ motifs, at intermediate 25 values not predicted by the extremes of the isolated models.
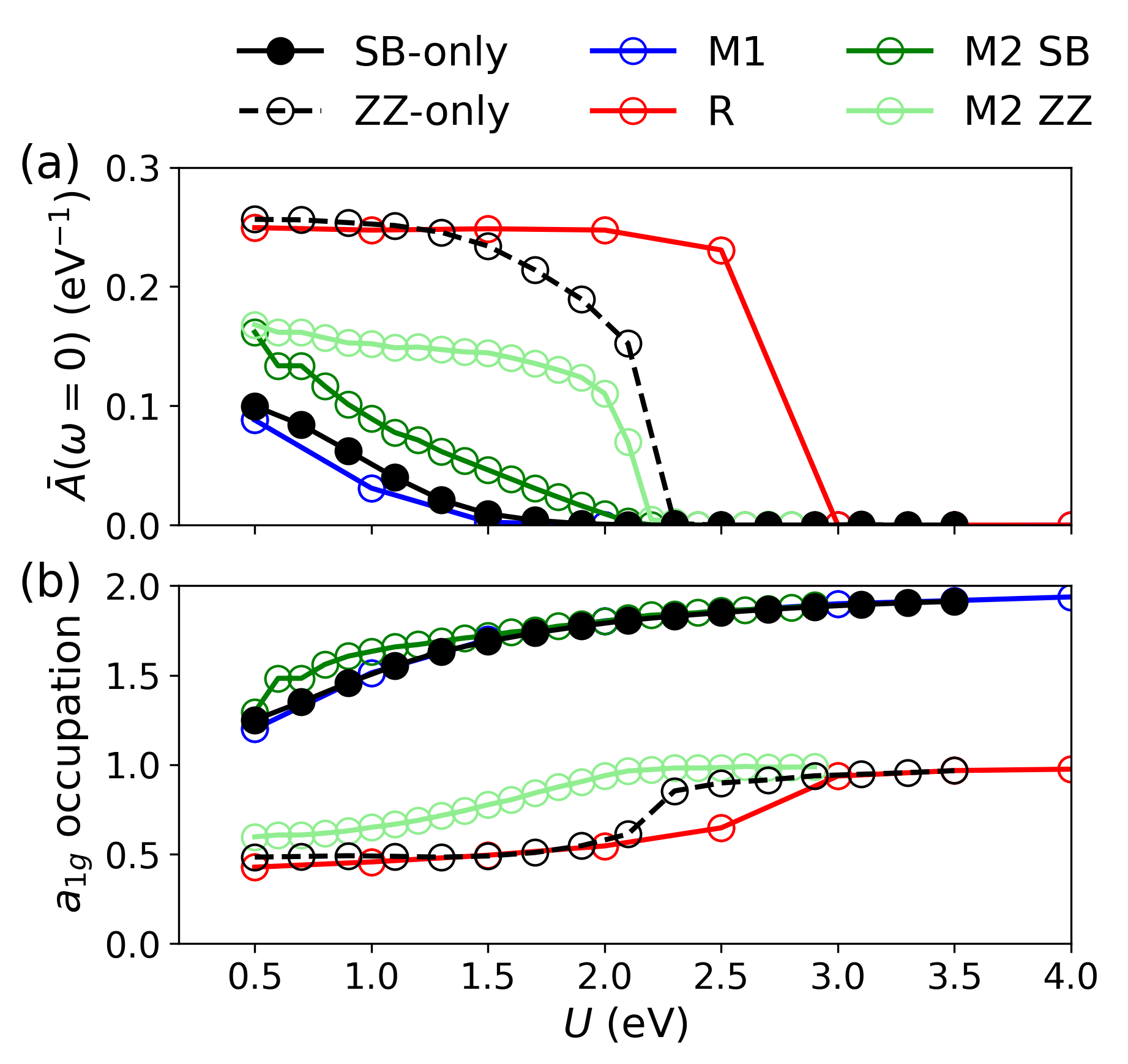
Figure 8: (a) Spectral weight and (b) 26 occupation as a function of 27 for R, M1, M2, SB-only, and ZZ-only structures.
Implications and Future Directions
The demonstrated ability of the bond-centered DFT+DMFT methodology to account for the dual character of electronic localization in M2, the energetics of all key phases, and the precise sequence of structural-electronic transitions highlights its suitability for general studies of complex transition metal oxides. This framework directly enables robust, chemically and symmetry-agnostic exploration of phase boundaries, the influence of strain (and thus heterostructure engineering), chemical substitutions, or defect-induced modifications—parameters of direct relevance for device applications hinging on MIT control.
The observation that unit cell strain is essential for M2 stability has implications for epitaxial growth strategies and strain-engineered MIT tuning. Furthermore, the approach is poised for extension to other systems where coexisting and intertwined Peierls/Mott physics operates, and for exploring the influence of disorder, vacancies, or nonequilibrium driving by external fields.
Conclusion
This work establishes the bond-centered DFT+DMFT approach as a theoretically sound, computationally efficient, and physically transparent method for unified studies of correlated-structural transitions in VO28. It reconciles the dual Peierls/Mott behaviors in a single correlated framework across all major structural forms without ad hoc cluster partitioning. The findings clarify the nature of the M2 and T phases, underscore the importance of lattice strain in MIT energetics, and enable targeted future investigations of control parameters essential to the functional utilization of VO29 and related materials.
(2603.26452)