A Pre-trained Reaction Embedding Descriptor Capturing Bond Transformation Patterns
Abstract: With the rise of data-driven reaction prediction models, effective reaction descriptors are crucial for bridging the gap between real-world chemistry and digital representations. However, general-purpose, reaction-wise descriptors remain scarce. This study introduces RXNEmb, a novel reaction-level descriptor derived from RXNGraphormer, a model pre-trained to distinguish real reactions from fictitious ones with erroneous bond changes, thereby learning intrinsic bond formation and cleavage patterns. We demonstrate its utility by data-driven re-clustering of the USPTO-50k dataset, yielding a classification that more directly reflects bond-change similarities than rule-based categories. Combined with dimensionality reduction, RXNEmb enables visualization of reaction space diversity. Furthermore, attention weight analysis reveals the model's focus on chemically critical sites, providing mechanistic insight. RXNEmb serves as a powerful, interpretable tool for reaction fingerprinting and analysis, paving the way for more data-centric approaches in reaction analysis and discovery.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
Overview: What this paper is about
This paper introduces RXNEmb, a “reaction fingerprint” created by a computer model to describe chemical reactions. Instead of relying on human-made rules, the model learns patterns directly from data—especially how chemical bonds break and form. The goal is to help scientists group, search, and understand reactions in a more accurate and flexible way.
What questions the researchers wanted to answer
- Can a computer learn the basic “bond change” patterns that define chemical reactions, just by looking at lots of examples?
- If we turn each reaction into a learned fingerprint (RXNEmb), can we:
- Group similar reactions together in a data-driven way?
- Draw simple maps that show how different reaction datasets compare?
- Peek inside the model to see which atoms it thinks are most important in a reaction?
How the researchers approached the problem
Think of reactions like short stories: molecules (characters) start in one situation (reactants) and end in another (products), with key “plot points” being bonds that break or form.
Here’s the approach in everyday terms:
- Learning from real vs. fake reactions:
- The team collected about 6.8 million real reactions and made a similar number of “fake” ones by messing up the products (like building Lego models with pieces that don’t fit).
- The model was trained to tell real from fake. To do that well, it had to learn what valid bond changes look like.
- Turning reactions into fingerprints:
- The model reads both the starting mixture (reactants, solvents, additives) and the products.
- Inside, it uses two tools:
- A graph neural network (GNN): treats a molecule like a network of atoms connected by bonds—like reading a map of cities (atoms) and roads (bonds).
- A Transformer: a type of model that uses “attention” to focus on the most important parts—like highlighting key sentences in a paragraph.
- It creates a single fixed-length vector (a fingerprint) that captures the overall bond changes. This fingerprint is called RXNEmb.
- Using RXNEmb for analysis:
- Clustering: Grouping reactions that have similar RXNEmb fingerprints.
- Visualization: Using a method called UMAP to compress these fingerprints into 2D, so we can plot and compare reaction spaces.
- Interpretability: Looking at the model’s “attention” to see which atoms it thinks are most important (usually the ones where bonds change).
What they found and why it matters
- Data-driven reaction grouping matches chemistry logic:
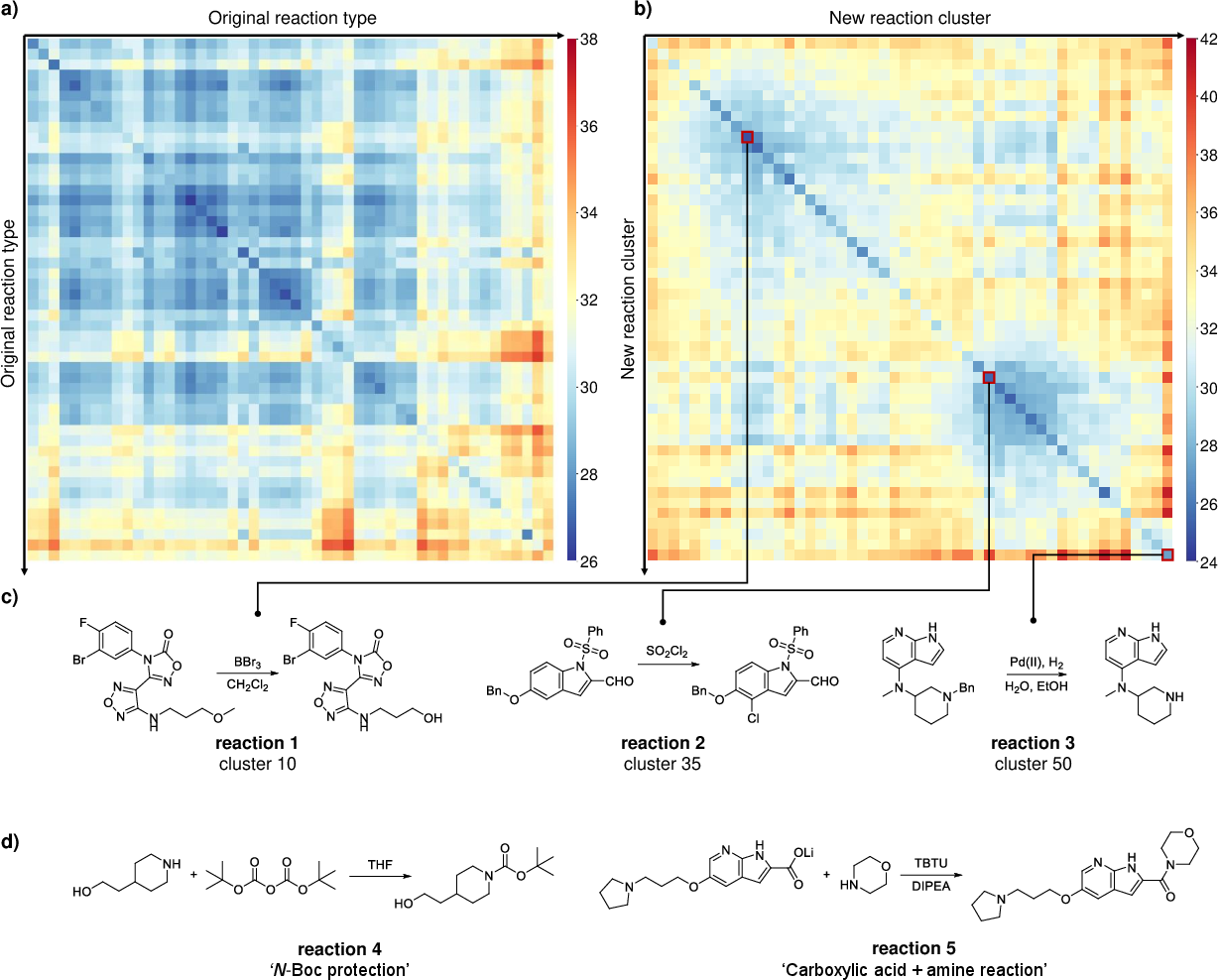
- Using RXNEmb, the team reclustered a popular dataset (USPTO-50k) without relying on expert-made labels.
- Reactions that involve similar bond changes were grouped together—even when old labels put them in different categories.
- Example: Two reactions that both form an amide bond were grouped together, even though they had different original labels. That’s useful because bond changes are often what chemists care about most.
- Better “maps” of reaction space:
- When plotting RXNEmb for multiple datasets, a large general dataset spread widely (many kinds of reactions), while specialized benchmark datasets clustered tightly (few bond-change types).
- This gives a quick visual check of how diverse a dataset is. It’s like seeing whether your music library is all one genre or covers many styles.
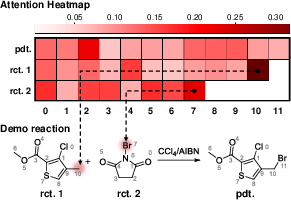
- The model focuses on the right atoms:
- By visualizing attention, the model highlighted atoms directly involved in bond changes (like the carbon that gets brominated and the bromine atom in the reagent).
- This is helpful because it shows the model is “thinking” in a chemically sensible way, which builds trust.
Why this work is useful
- Smarter reaction search and grouping:
- RXNEmb lets you find similar reactions by their bond-change patterns, which is great for discovery, literature mining, or picking training data.
- Clearer dataset planning:
- The 2D maps help researchers see whether a dataset has enough diversity for a given study or if it’s too narrow.
- Interpretable AI for chemistry:
- Attention maps show where the model is “looking,” aligning with how chemists reason about mechanisms.
- Practical limitations and good use cases:
- RXNEmb is best at capturing bond changes between reactants and products. It doesn’t directly encode subtle effects from catalysts or ligands (which can control reactivity and selectivity).
- Still, it’s very useful for:
- Few-shot learning: quickly finding similar reactions to boost small datasets.
- Transfer learning: fine-tuning the model for specific tasks after it has learned general reaction knowledge.
Bottom line: What this could change
RXNEmb provides a general, learned “fingerprint” for reactions that:
- Reflects real chemical logic (bond making and breaking),
- Helps cluster and visualize reactions without hand-made rules,
- And offers interpretable insights into what the model considers important.
As more reaction data becomes available, tools like RXNEmb can make reaction analysis faster, more consistent, and more data-driven—supporting better prediction, smarter experiment planning, and potentially quicker discovery in chemistry.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a consolidated list of what remains missing, uncertain, or unexplored in the paper, framed to be concrete and actionable for future research.
- Quantitative validation that RXNEmb captures bond-change patterns is absent: assess correlations between embedding distances and explicit bond-change metrics (e.g., number/type of formed/cleaved bonds), and benchmark classification accuracy against curated mechanism labels.
- No comparison against established reaction descriptors (e.g., DRFP, rxnfp, CGR-based fingerprints): run head-to-head evaluations on reaction similarity, clustering validity, and downstream predictive tasks.
- Cluster formation is under-specified and unvalidated: the choice of 50 clusters is arbitrary; evaluate optimal cluster numbers (silhouette, Davies–Bouldin), cluster stability across seeds/hyperparameters, and external validity against mechanistic taxonomies.
- Distance metric for embedding similarity is not reported: compare Euclidean, cosine, and learned Mahalanobis distances; calibrate distance thresholds for “similar reactions” with domain-grounded criteria.
- UMAP visualization is qualitative only: perform sensitivity analysis to UMAP hyperparameters and seeds; complement with quantitative diversity/coverage metrics (e.g., entropy, dispersion, neighborhood overlap) and alternative projections (PCA, t-SNE).
- Attention-based interpretability is demonstrated on a single case: conduct large-scale, quantitative attribution studies (attention, gradients, integrated gradients) across diverse reactions to verify alignment with known reactive centers and bond-change sites.
- The pretraining “fictitious reaction” generation (random product fragment exchange) may introduce biases: test chemically plausible decoys (stoichiometry-preserving, condition-aware, atom-mapping-consistent negatives) and quantify robustness to decoy design.
- Label noise in “real” reactions and possible plausibility of some “fictitious” products are not addressed: estimate noise rates, apply robust training (e.g., noise-aware loss, bootstrapping), and measure sensitivity to mislabeled data.
- Reaction conditions, catalysts, and ligands are not explicitly modeled in RXNEmb (beyond inclusion as molecules): quantify the descriptor’s sensitivity to condition/catalyst changes and explore condition-aware embeddings to capture SPR/ligand effects.
- Lack of ablation studies on architecture choices: evaluate contributions of separate reactant/product encoders, difference+concatenation scheme, Jumping Knowledge, attention pooling, and Transformer depth to descriptor quality.
- Invariance to reagent ordering and stoichiometry is not demonstrated: prove or enforce permutation invariance and test robustness with shuffled/multiplicity-varied components and multi-product reactions.
- Handling of stereochemistry and 3D effects is unclear: benchmark RXNEmb on stereochemical outcomes (enantio-/diastereoselectivity), and explore 3D-informed or conformer-augmented models to capture subtle geometric effects.
- Coverage and curation of the pretraining dataset are insufficiently detailed: publish dataset statistics (reaction type distribution, condition diversity), sources, cleaning steps, and bias assessments; release a representative subset if full data cannot be shared.
- Scalability and efficiency are not quantified: report memory/time costs for encoding and clustering at scale, and explore batching, model compression, and approximate nearest neighbor indexing for large databases.
- Generalization beyond USPTO-50k is not demonstrated: evaluate on diverse domains (organometallics, inorganic, polymerizations, biocatalysis, multi-step/telescoped reactions) to assess breadth and limits of RXNEmb.
- Uncertainty quantification and confidence measures are missing: develop calibrated similarity scores or probabilistic embeddings to express confidence in reaction similarity/retrieval outcomes.
- Mechanistic discrimination (e.g., SN1 vs SN2, E1 vs E2) is untested: create controlled benchmark sets to test whether RXNEmb separates mechanistically distinct pathways with similar substrates/products.
- Reproducibility gaps: pre-trained RXNGraphormer weights and full pretraining data are not provided; release weights/checkpoints and training recipes to enable independent validation.
- Integration into downstream workflows is only suggested: demonstrate retrieval-augmented modeling, active learning loops, and end-to-end pipelines showing measurable gains in reaction prediction/selectivity or synthesis planning.
- Similarity-based retrieval effectiveness is not evaluated: measure precision/recall/mean average precision for retrieving truly analogous reactions and assess utility in experimental design or data augmentation.
- Robustness to representation choices is untested: verify invariance to SMILES canonicalization/aromaticity normalization, atom-mapping variations, and missing/incorrect reagents; test graph vs SMILES input pathways.
- Mapping RXNEmb clusters to human-interpretable taxonomies is not developed: build automatic cluster labeling (e.g., via bond-change rule extraction), and evaluate alignment with expert taxonomies (NameRxn, RDChiral classes).
- Fine-tuning and transfer learning protocols are not characterized: specify data requirements, gains over training-from-scratch baselines, and failure modes for tasks emphasizing catalyst/ligand-driven outcomes.
- Treatment of charged species, metals, and coordination chemistry is unclear: expand atom/edge features to better represent organometallic/inorganic reactions and evaluate descriptor performance on metal-mediated transformations.
- Effect of noisy or incomplete reaction records (e.g., missing solvents/additives, ambiguous products) is not studied: quantify performance degradation and propose data cleaning or robustness strategies.
Practical Applications
Below is a structured mapping from the paper’s findings to concrete applications across sectors, with time-to-deploy assessments, potential tools/workflows, and key assumptions or dependencies.
Immediate Applications
- Bold, chemistry-native reaction fingerprinting for retrieval and clustering
- Sectors: pharmaceuticals, specialty chemicals, materials; software/cheminformatics
- Tools/workflows: RXNEmb indexing and kNN/ANN search; ELN/LIMS plugins for “similar reaction” lookup; de-duplication and near-duplicate detection; cluster-based navigation of corporate reaction databases
- Assumptions/dependencies: Access to the RXNEmb model and code; SMILES standardization/canonicalization; data governance/privacy; embeddings capture bond-change similarity but not detailed conditions
- Data-driven reaction taxonomy to replace/augment rule-based labeling
- Sectors: database curation (Reaxys/CAS), ELNs, academia
- Tools/workflows: Automated re-clustering pipelines for USPTO/corporate datasets; crosswalks from RXNEmb clusters to legacy taxonomies (e.g., NameRxn); dashboarding of cluster heatmaps
- Assumptions/dependencies: Agreement on desired granularity; acceptance of data-driven taxonomies; periodic recalibration when scope expands
- Reaction space diversity and bias audits for dataset design
- Sectors: pharma/materials R&D, ML for chemistry (academia/industry)
- Tools/workflows: UMAP/2D maps over RXNEmb; dataset coverage KPIs; selection of training/validation splits reflecting space coverage; active learning seed selection from sparse clusters
- Assumptions/dependencies: Sufficient dataset size; reproducible visualization parameters; recognition that RXNEmb emphasizes bond-change diversity not catalyst/condition diversity
- Similarity-based data augmentation and few-shot baselines
- Sectors: pharma/materials (HTE, medchem); ML platform teams
- Tools/workflows: Retrieval-augmented training (nearest-neighbor augmentation); similarity-weighted loss functions; kNN predictors using RXNEmb as a fast baseline
- Assumptions/dependencies: Locality in RXNEmb correlates with transferability; careful OOD detection to avoid negative transfer
- Plausibility filtering for forward prediction and retrosynthesis
- Sectors: cheminformatics software vendors; internal tools in R&D IT
- Tools/workflows: RXNEmb-based real/fictitious plausibility score as a filter or re-ranking term in route planners; early triage of invalid reactions generated by template-free models
- Assumptions/dependencies: Threshold calibration per chemistry domain; integration with existing planners; monitoring for domain shift
- Mechanistic insight and QC via attention visualization
- Sectors: academia; process and discovery chemists in industry
- Tools/workflows: Attention-map viewers to highlight reactive centers; rapid sanity-checks for atom mappings and reaction SMILES; hypothesis generation for site selectivity
- Assumptions/dependencies: Attention ≠ causality; usefulness improves with correct inputs and consistent preprocessing
- Rapid prior-art triage and bond-change–centric patent search
- Sectors: IP analytics, legal; competitive intelligence
- Tools/workflows: Clustered patent reaction corpora; white-space scans by bond-change type; novelty checks for proposed transformations
- Assumptions/dependencies: Reliable reaction extraction (OCR/NLP) and SMILES normalization from patents; legal/compliance review
- Curriculum aids and interactive teaching of reaction space
- Sectors: education (undergrad/graduate/continuing), edtech
- Tools/workflows: Interactive maps showing clusters and exemplars; teaching modules contrasting rule-based vs data-driven classifications
- Assumptions/dependencies: Simple UI; curated example sets for pedagogy
- Data quality control and curation accelerators
- Sectors: data engineering in R&D IT, database providers
- Tools/workflows: Outlier and mislabeled-class detection via cluster assignment; duplicate/near-duplicate detection; automated flags for inconsistent reactant/product pairs
- Assumptions/dependencies: Clear curation policies; human-in-the-loop review
Long-Term Applications
- Fine-tuned predictors for yield/selectivity integrated with lab automation
- Sectors: pharmaceuticals, agrochemicals, materials
- Tools/workflows: RXNGraphormer fine-tuned on HTE datasets; closed-loop optimization (planner + robot + analytics); design-of-experiments accelerators guided by RXNEmb similarity
- Assumptions/dependencies: High-quality labeled condition/yield/selectivity data; robust lab-automation integration; careful generalization beyond bond-change patterns
- Autonomous synthesis planning with RXNEmb-aware scoring
- Sectors: software/robotics for autonomous labs
- Tools/workflows: Multi-objective retrosynthesis and forward search using plausibility and similarity priors; on-the-fly feasibility checks to prune search trees
- Assumptions/dependencies: Scalable search infrastructure; coupling to condition recommendation and cost/safety models
- Reaction recommendation copilots in ELNs/LIMS
- Sectors: pharma/materials R&D; academia
- Tools/workflows: Contextual suggestions of transformations and precedent reactions; “you might try” panels driven by bond-change clusters; link-outs to conditions literature
- Assumptions/dependencies: Condition inference models and metadata; IP/privacy controls; UX integration and user trust
- Standardization and policy for reaction data reporting
- Sectors: policy/regulatory, standards bodies, journals
- Tools/workflows: Guidelines to include reaction-level descriptors (e.g., bond-change fingerprints) in submissions; dataset diversity audits as a reporting standard
- Assumptions/dependencies: Community consensus; alignment with FAIR data principles; incentives for adoption
- Green chemistry and process-optimization analytics
- Sectors: process chemistry, energy/materials manufacturing
- Tools/workflows: Route scoring by avoidable bond-change classes (e.g., deprotections, halogenations); mapping to greener alternatives within nearby clusters; E-factor/LCA overlays
- Assumptions/dependencies: Integration with LCA/toxicity databases; organizational targets for green metrics
- Patent landscaping and strategic whitespace discovery at scale
- Sectors: finance, corporate strategy, IP
- Tools/workflows: Macro-level maps of transformation space across assignees; detection of underexplored bond-change domains; M&A scouting for complementary portfolios
- Assumptions/dependencies: Comprehensive patent corpora; reliable entity resolution; legal review
- Cross-modal chemistry foundation models
- Sectors: software/AI; healthcare/materials
- Tools/workflows: Joint embeddings linking RXNEmb to molecular properties, spectra, or text; unified retrieval and reasoning across reactions, molecules, and conditions
- Assumptions/dependencies: Large, aligned multimodal datasets; compute and MLOps maturity
- Safety and hazard screening in autonomous platforms
- Sectors: robotics, EHS, process safety
- Tools/workflows: Pre-execution safety filters tagging hazardous bond-change patterns; integration with kinetic/thermo risk models and reagent incompatibility databases
- Assumptions/dependencies: Curated safety labels; calibration on near-miss and incident data
- Adaptive, data-driven chemistry education platforms
- Sectors: edtech, universities
- Tools/workflows: Personalized curricula that traverse reaction space from fundamentals to advanced clusters; assessment by ability to generalize across nearby bond-change classes
- Assumptions/dependencies: Content development; instructor adoption; learning analytics
- Market-grade APIs and SaaS for reaction similarity and clustering
- Sectors: software/SaaS providers, CROs
- Tools/workflows: Managed RXNEmb services (vector databases, search, clustering, dashboards); enterprise connectors for ELNs and data lakes
- Assumptions/dependencies: Business models for usage-based pricing; SLAs, data security certifications
- Discovery in energy/materials (electrolytes, polymers, catalysts)
- Sectors: energy storage, performance materials
- Tools/workflows: Map known transformations for polymer backbones/functionalizations; suggest analogous bond-change routes to tune properties; transfer learning from adjacent clusters
- Assumptions/dependencies: Domain-specific property labels; bridging from transformation similarity to macroscopic performance
Notes on overarching feasibility:
- RXNEmb prioritizes bond formation/cleavage patterns; condition, catalyst, and ligand effects may require fine-tuning or complementary models.
- Data quality (clean SMILES, correct reactant/product sets, robust preprocessing) is critical.
- Domain shift is real: embeddings trained on public data may need adaptation for proprietary or niche chemistries.
- Interpretability is helpful (attention maps) but should be validated experimentally; attention is indicative, not definitive causality.
Glossary
- Attention weight analysis: A method of interpreting which parts of a model receive higher attention, highlighting important features or sites. "attention weight analysis reveals the model's focus on chemically critical sites, providing mechanistic insight."
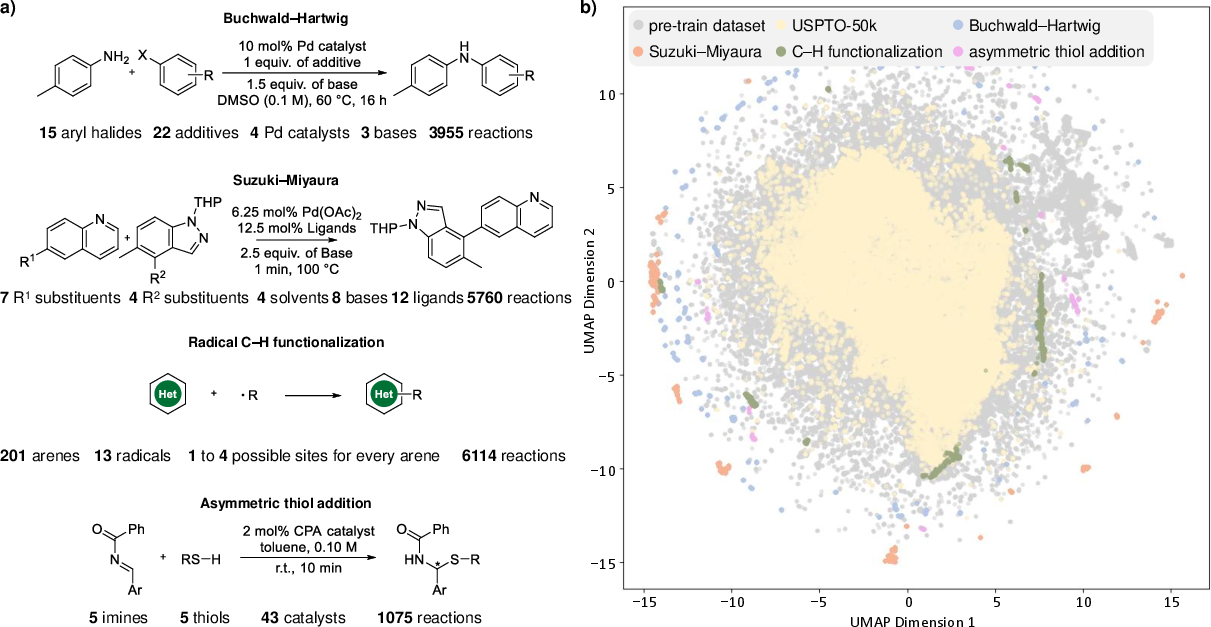
- BuchwaldâHartwig coupling: A palladium-catalyzed cross-coupling forming C–N bonds between aryl halides and amines. "BuchwaldâHartwig coupling"
- Contrastive learning: A training paradigm that learns by distinguishing between positive and negative pairs, enhancing representation quality. "By performing contrastive learning on this large-scale dataset to distinguish between real and fictitious reactions, the model intrinsically learns the correct patterns of chemical bond changes."
- Differential reaction fingerprint (DRFP): A reaction descriptor computed from the symmetric difference of substructures between reactants and products. "A representative example is the differential reaction fingerprint (DRFP) developed by Reymond"
- Dimensionality reduction: Techniques that project high-dimensional data into lower dimensions for visualization or analysis. "Combined with dimensionality reduction, RXNEmb enables visualization of reaction space diversity."
- Fictitious reactions: Artificial reactions created to violate chemical rules, used for training or evaluation. "an equal number of fictitious reactions constructed by randomly generating fictitious products via fragment exchange on the SMILES of real products."
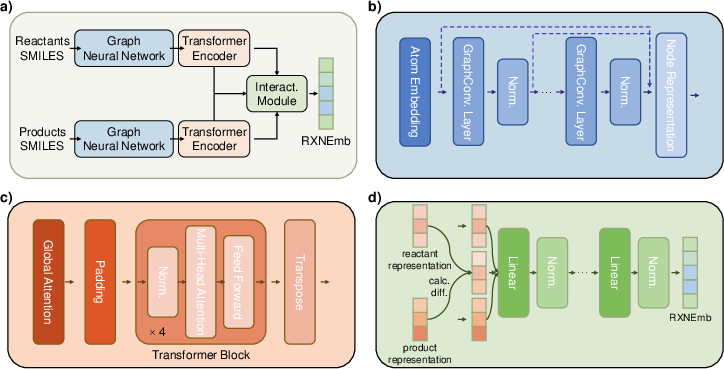
- Global attention-based pooling: A pooling mechanism that weights nodes (e.g., atoms) by learned attention to form a graph-level representation. "via a global attention-based pooling layer"
- Graph convolutional layers: Neural network layers that perform convolution operations over graph structures (nodes/edges). "The GNN for encoding a single molecule consists of 4 graph convolutional layers"
- Graph neural network (GNN): A neural architecture designed to operate on graph-structured data, such as molecular graphs. "each encoder set contains a graph neural network (GNN) for encoding individual molecules"
- Jumping Knowledge: A technique to aggregate representations from multiple GNN layers to mitigate over-smoothing. "Jumping Knowledge connections are employed to aggregate information from multiple layers"
- Kennard-Stone (KS) algorithm: A sampling method that selects representative, diverse points in descriptor space. "We first used the Kennard-Stone (KS) algorithm to select 50 representative reactions that are distant from each other in the descriptor space as initial centroids."
- Mechanistic insight: Understanding of the underlying chemical mechanism or bond-change processes. "providing mechanistic insight."
- NameRxn: Software for rule-based classification of reactions into named types. "which were originally classified by the NameRxn software"
- N-Bromosuccinimide (NBS): A brominating reagent used for selective bromination reactions. "the brominating reagent N-Bromosuccinimide (NBS, rct. 2)"
- Optimal leaf ordering: A method to reorder clustered categories to make heatmaps or dendrograms more interpretable. "Furthermore, optimal leaf ordering was applied to the clustering result, ensuring that categories close in the latent space also have adjacent indices in the sorted order."
- Reaction fingerprinting: Creating fixed-length descriptors that capture reaction characteristics for comparison or retrieval. "RXNEmb serves as a powerful, interpretable tool for reaction fingerprinting and analysis"
- Reaction space: The conceptual or embedded landscape of reactions characterized by their descriptors. "RXNEmb enables visualization of reaction space diversity."
- RXNEmb: A pre-trained reaction-level embedding capturing bond formation and cleavage patterns. "This study introduces RXNEmb, a novel reaction-level descriptor derived from RXNGraphormer"
- RXNGraphormer: A pre-trained deep learning framework for reactions that learns bond-change patterns via real/fictitious discrimination. "we developed RXNGraphormer, a pre-trained deep learning framework applicable to reaction performance prediction and synthesis planning."
- rxnfp: A data-driven reaction descriptor learned from a reaction classification model. "the rxnfp descriptor developed by Reymond and Schwaller"
- Self-attention: A mechanism in Transformers that relates different positions within a sequence to compute representations. "which utilizes multi-head self-attention to effectively capture interaction information among all molecules within the reaction system"
- SMILES: A textual notation system for representing molecular structures. "represented as SMILES strings"
- Sonogashira coupling: A cross-coupling between aryl/alkenyl halides and terminal alkynes, typically palladium-catalyzed. "Sonogashira coupling"
- Sterimol parameters: Steric descriptors that quantify substituent dimensions along defined axes. "Sterimol parameters"
- Stille reaction: A palladium-catalyzed cross-coupling using organostannanes to form C–C bonds. "Stille reaction"
- SuzukiâMiyaura coupling: A palladium-catalyzed cross-coupling of boronic acids with aryl/alkenyl halides to form C–C bonds. "SuzukiâMiyaura coupling"
- UMAP: Uniform Manifold Approximation and Projection, a non-linear dimensionality reduction method for visualization. "We then employed UMAP to co-project the reaction features from all these datasets"
- USPTO-50k: A benchmark dataset of 50,000 reactions extracted from USPTO records. "USPTO-50k dataset"
- Wohl-Ziegler bromination: A selective benzylic or allylic bromination method using NBS under radical conditions. "Taking the concrete example of a Wohl-Ziegler bromination reaction"
Collections
Sign up for free to add this paper to one or more collections.