- The paper presents an end-to-end quantum workflow that transforms 3D-RISM water densities into a QUBO problem for hydration-site prediction.
- The quantum approach, executed on IBM Heron processors, outperforms classical methods in sampling success and optimization efficiency.
- Resource scaling forecasts indicate a practical quantum advantage for drug discovery with hardware improvements expected by 2028.
Quantum Optimization for Protein Pocket Hydration-Site Prediction
Introduction
The accurate prediction of protein pocket hydration sites is a persistent computational and methodological bottleneck in drug discovery pipelines. Water molecules profoundly influence protein conformational dynamics, binding thermodynamics, and ligand recognition—necessitating precise placement of hydration sites for binding free energy calculations and lead optimization. Traditional techniques—including molecular dynamics (MD), Monte Carlo sampling, and hybrid methods like 3D-RISM—remain resource-intensive and can struggle with sampling dense water networks, even on HPC infrastructure. Recently, quantum computing has emerged as an alternative paradigm, offering theoretical speedups for combinatorial and sampling problems mapped onto Ising-like Hamiltonians.
This paper (2512.08390) presents an end-to-end quantum computing workflow for hydration-site prediction in realistic protein-ligand complexes. It integrates classical computation of 3D-RISM water densities, formulation of the discrete water placement task as a Quadratic Unconstrained Binary Optimization (QUBO) problem, and high-fidelity quantum hardware execution using Q-CTRL's hybrid optimization stack on IBM's Heron processors. The approach is systematically benchmarked on health-relevant proteins, offering numerical evidence for utility at near-term scales and providing detailed resource forecasts for quantum advantage.
Methodology and Workflow Architecture
The hydration-site prediction pipeline initiates from the computation of 3D-RISM water densities (g(r)), leveraging AMBER tools and standardized force fields. This continuous density is discretized via Gaussian Mixture Modeling (GMM), with a dimensionality reduction step applied to ensure problem feasibility on quantum hardware. This step involves grid resampling and density thresholding (δ, τg), balancing site enumeration against device constraints.
A binary variable grid is defined over the reduced region, mapping each candidate site to a QUBO variable. The cost function encodes not only the energy landscape but also essential spatial exclusion constraints via pairwise quadratic penalties. The QUBO formalism is cast as:
C(x)=i=1∑NQiixi+2i<j∑NQijxixj
where xi is binary and Qij encodes interaction and exclusion terms. The full-stack conversion—from 3D-RISM to QUBO and input to a quantum variational optimization algorithm—is illustrated in the workflow schematic.
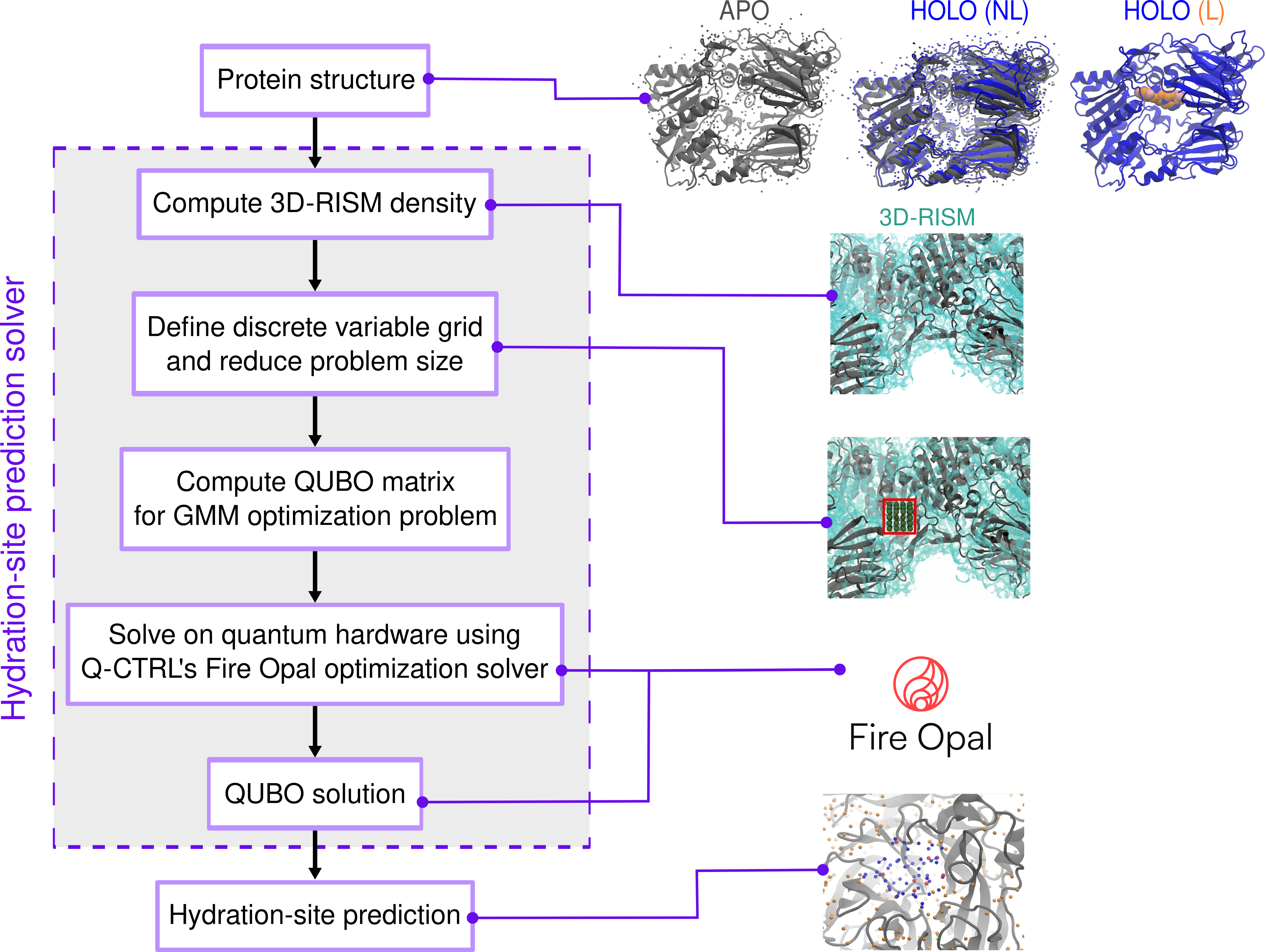
Figure 1: Full-stack integration from continuous 3D-RISM density to QUBO formulation and quantum algorithmic prediction of hydration sites.
Quantum execution is performed using Q-CTRL's Fire Opal, incorporating automated error suppression and postprocessing for robust optimization on NISQ devices. The QAOA-like ansatz is compiled for the specific device topology and run on latest IBM Heron backends (up to 156 qubits per instance).
Quantum Hardware Benchmarks and Comparative Results
A suite of protein-ligand complexes, including FDA-approved drug targets, were used to validate the workflow. QUBO instance sizes ranged up to 123 variables (qubits), forced by hardware limits. The quantum solver demonstrated high success probabilities in identifying optimal or near-optimal solutions, outperforming greedy local solvers and matching simulated annealing (SA) in smaller problem instances.
For instance "d" (116 variables), the quantum solver sampled the global optimum at 9% probability compared to 2% for SA:
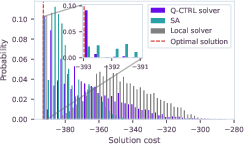
Figure 2: Cost distributions for the QUBO instance "d" showing quantum output versus SA and local greedy approaches.
Global benchmarking across the test set confirmed consistent quantum solution quality:
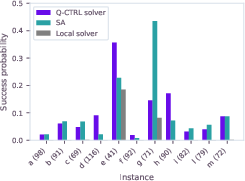
Figure 3: Success probabilities for optimal solution found by Q-CTRL solver, SA, and local greedy approaches across protein-ligand systems.
Notably, for the largest tractable instance (3b7e with ligand, 123 variables), exact classical solvers stalled with a 40% optimality gap at the time cut-off, while quantum optimization reached lower costs in 25 minutes, providing a strong empirical indication of practical quantum utility for instances where classical exact solvers fail.
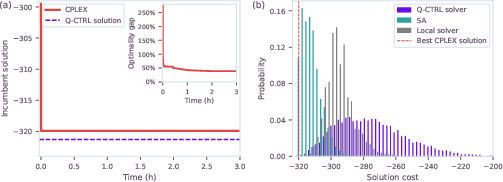
Figure 4: Quantum versus classical solution trajectories and cost distributions for a large hydration-site QUBO instance (123 variables).
Device scaling was explicitly benchmarked between IBM Heron r2 and r3 processors, with the latter displaying superior sampling probabilities, highlighting rapid hardware evolution and its impact on quantum workflow efficacy.
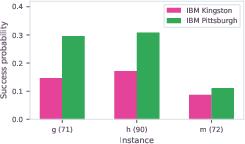
Figure 5: Comparative quantum solver success rates on Heron r2 (IBM Kingston) and r3 (IBM Pittsburgh) for selected hydration-site prediction instances.
Prediction Accuracy, Problem Scaling, and Comparison with Classical Methods
Hydration-site prediction performance was quantitatively assessed using experimental crystal water (CW) positions as ground truth. Multiple metrics were applied, including fraction of CWs recovered (C), average cluster size (⟨CS⟩), and precision (P∗,⟨P⟩). QUBO instance size and grid granularity were systematically scaled to probe accuracy and robustness.
Precision and recovery grew monotonically with QUBO size. For the 123-variable instance, ∼60% of CWs were correctly identified, with increasing robustness in larger instances up to 900+ variables (classically evaluated) and beyond.
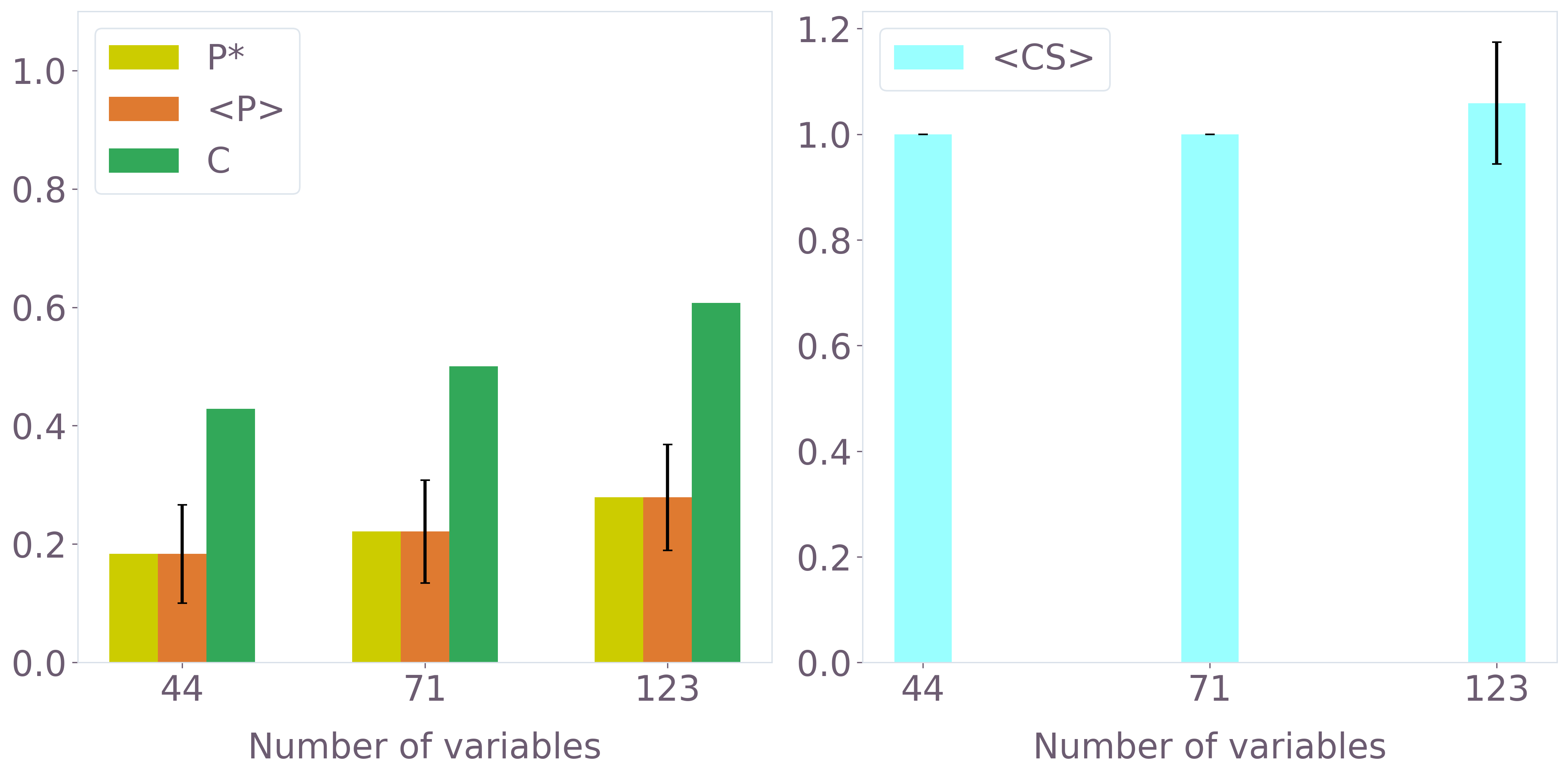
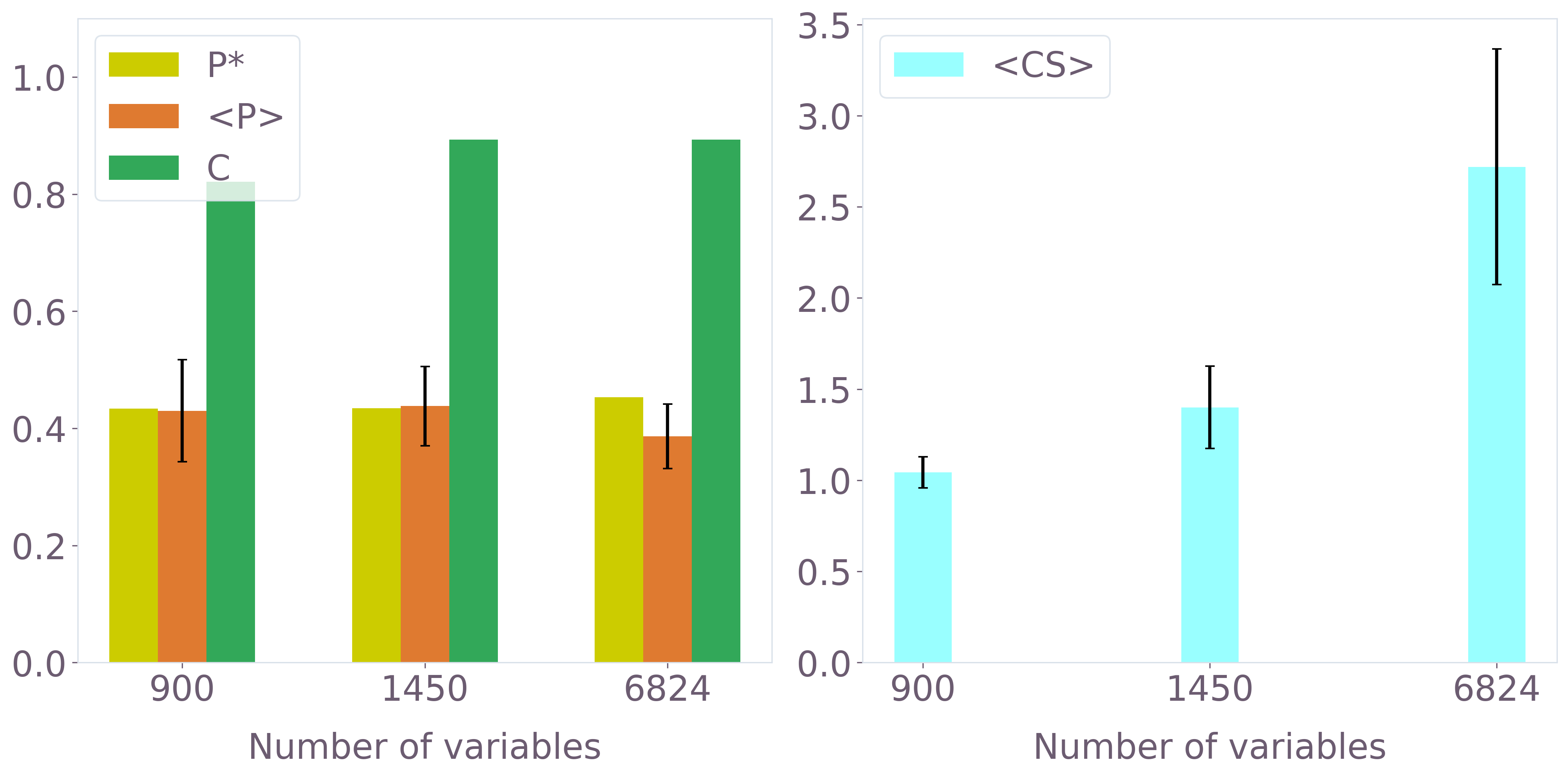
Figure 6: Hydration-site prediction metrics as a function of QUBO instance size for 3b7e with ligand (quantum and classical SA-driven optimization).
Spatial mapping of predicted waters demonstrated proximity to crystallographic locations and stability with increased variable count, visualized in both 3D and PCA-reduced 2D spaces.
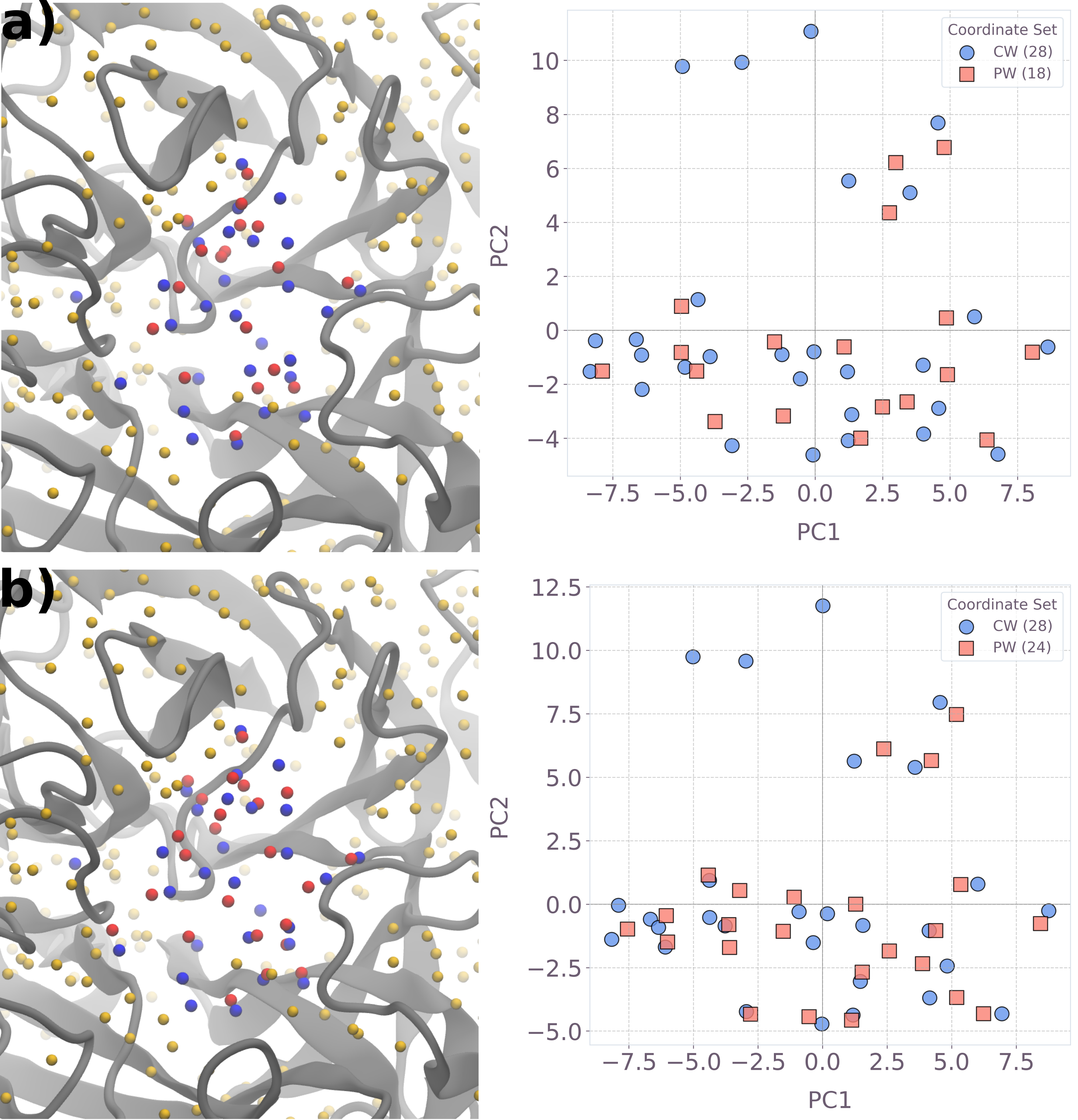
Figure 7: 3D and 2D PCA visualizations of hydration sites predicted by QUBO optimization for different instance sizes.
Comparison against established hydration-site prediction methods (Hydraprot, Placevent, Watgen, Dowser++) under identical test conditions indicated QUBO-based quantum optimization outperforms or matches top classical approaches in both precision and recovery. Notably, Placevent (which also consumes 3D-RISM input) was significantly inferior, underlining the advantage of discrete QUBO formulation and quantum exploration.
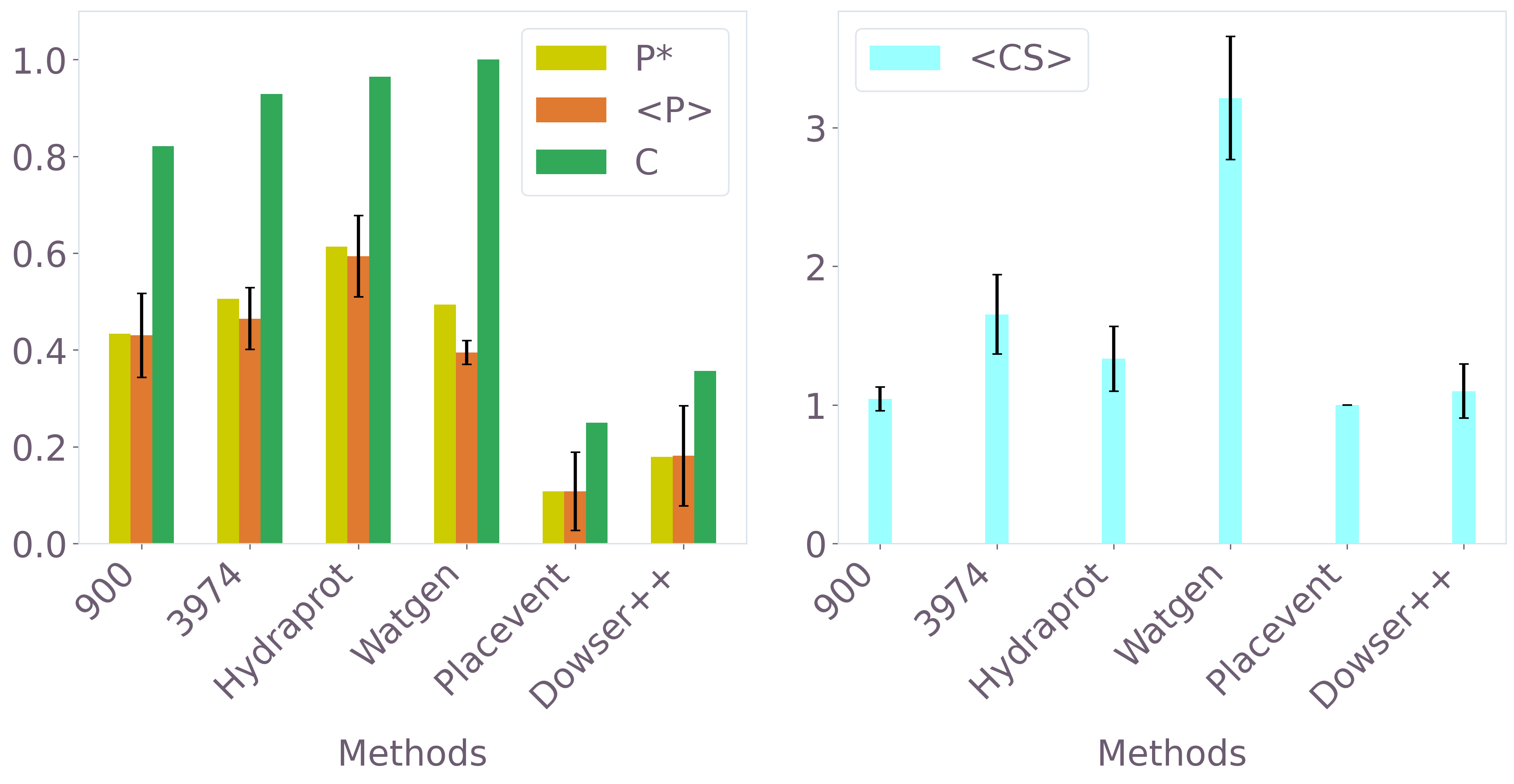
Figure 8: Comparative performance of QUBO-optimized predictions versus Hydraprot and other classical methods across standard hydration-site metrics.
Resource Scaling and Quantum Advantage Forecasting
Resource requirements were extrapolated based on two-qubit gate scaling with variable count; for instances competitive with leading methods (∼900 variables), gate counts approach 105, surpassing current error thresholds but within near-term reach pending error correction advances and hardware roadmap projections. Gate scaling was quadratic, strongly motivating development of compilation and connectivity solutions.
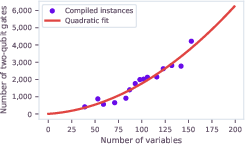
Figure 9: Two-qubit gate scaling for hydration-site QUBO instances, quadratic fit (red) suggests future feasibility at 1000-qubit devices with improved error correction.
According to IBM's publicly available roadmap, qubit counts around 1000 and requisite error-corrected gate depths are forecasted by 2027–2029, with quantum advantage over exact classical solvers (quantum utility) likely in selected use-cases by 2028.
Implications and Future Directions
This work sets a precedent for direct quantum computation of biochemically relevant structure–function properties in drug discovery. Quantum-enhanced hydration-site placement can meaningfully accelerate system preparation for MD or docking, and unlock combinatorially hard water network modeling at accuracy levels competitive with finest machine learning and physics-based approaches—at substantially reduced runtimes (once hardware scales up).
As hardware and algorithms continue evolving, similar QUBO workflows could extend to side-chain optimization [QAB_1], ligand docking [Ding_molecular_docking_2024], and full thermodynamic integration tasks using hybrid quantum-classical strategies. Practical deployment will require further advances in error correction, circuit depth management, and seamless hardware-software integration.
While present limitations force dimensionality reduction and classical fallback for large-scale prediction, empirical evidence here indicates utility for quantum hardware in the noisy intermediate scale regime, with broad applicability in CADD, protein–ligand modeling, and beyond.
Conclusion
This paper demonstrates a rigorous, fully-integrated quantum computing workflow for hydration-site prediction in realistic protein pockets. By casting the 3D-RISM-based problem into QUBO and deploying on state-of-the-art quantum hardware, it achieves accuracy matching leading classical methods for small-to-medium instances and offers clear resource and performance scaling trajectories for quantum advantage by the end of the decade. The approach provides immediate value for drug discovery practitioners seeking automated, high-precision solvation modeling and paves the way for broader quantum acceleration of molecular simulation in pharmaceutical and chemical research.