LSM-MS2: A Foundation Model Bridging Spectral Identification and Biological Interpretation
Abstract: A vast majority of mass spectrometry data remains uncharacterized, leaving much of its biological and chemical information untapped. Recent advances in machine learning have begun to address this gap, particularly for tasks such as spectral identification in tandem mass spectrometry data. Here, we present the latest generation of LSM-MS2, a large-scale deep learning foundation model trained on millions of spectra to learn a semantic chemical space. LSM-MS2 achieves state-of-the-art performance in spectral identification, improving on existing methods by 30% in accuracy of identifying challenging isomeric compounds, yielding 42% more correct identifications in complex biological samples, and maintaining robustness under low-concentration conditions. Furthermore, LSM-MS2 produces rich spectral embeddings that enable direct biological interpretation from minimal downstream data, successfully differentiating disease states and predicting clinical outcomes across diverse translational applications.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What this paper is about (big picture)
This paper introduces LSM-MS2, a powerful AI model that helps scientists make sense of complicated lab data called mass spectrometry. Think of mass spectrometry as a machine that breaks molecules into pieces and records the pattern of those pieces—like smashing a LEGO model and noting which bricks fall off. Those patterns (called “spectra”) are like fingerprints for molecules. The problem is that most of these fingerprints are still unlabeled, so we don’t know which molecules they belong to. LSM-MS2 learns from millions of these fingerprints to:
- Identify what molecules are present, and
- Help understand what’s going on biologically (like spotting disease patterns), even when we don’t have many labeled examples.
The main goals, in simple terms
The paper focuses on two big questions:
- Can this AI identify molecules from their “broken piece” patterns more accurately than existing methods, especially when molecules look very similar (isomers)?
- Can the AI’s learned “map” of spectra be used to spot meaningful biological differences—like different diseases—without needing the usual long, manual steps of labeling everything first?
How the researchers did it (methods made simple)
- Training the model:
- LSM-MS2 is a “transformer” model (the same kind of AI used in chatbots). Instead of learning language, it learns the “language of molecules” from millions of spectra.
- It turns each spectrum (the pattern of broken pieces) into a compact vector called an embedding. You can think of this as placing each spectrum at a point on a giant map where similar molecules end up close together.
- Testing the model:
- Spectral identification (the “matching game”): Given an unknown spectrum, the model looks for the closest matches in a large library of known spectra (1.8 million spectra for 99,000 molecules). This is like trying to find which song you’re hearing by comparing it to a huge playlist of known songs.
- Biological interpretation (the “story behind the data”): Instead of labeling every molecule first, they combine all the spectrum-embeddings from a sample (like a patient’s blood sample) into a single sample-level embedding. Then simple machine learning tools use that to tell groups apart (for example, disease vs. healthy), much faster than traditional workflows.
- What they compared against:
- “Cosine similarity” (a classic way to compare spectra directly).
- DreaMS (a recent top deep learning model for spectra).
- How they measured success:
- Top-K accuracy: how often the correct molecule is in the top 1 or top 5 guesses.
- Structural closeness when the guess is wrong (MCES): are the mistakes at least chemically close?
- Real-world tests: performance under challenging conditions—like very similar molecules (isomers), messy biological samples, and very low concentrations.
What they found and why it matters
- Better molecule identification:
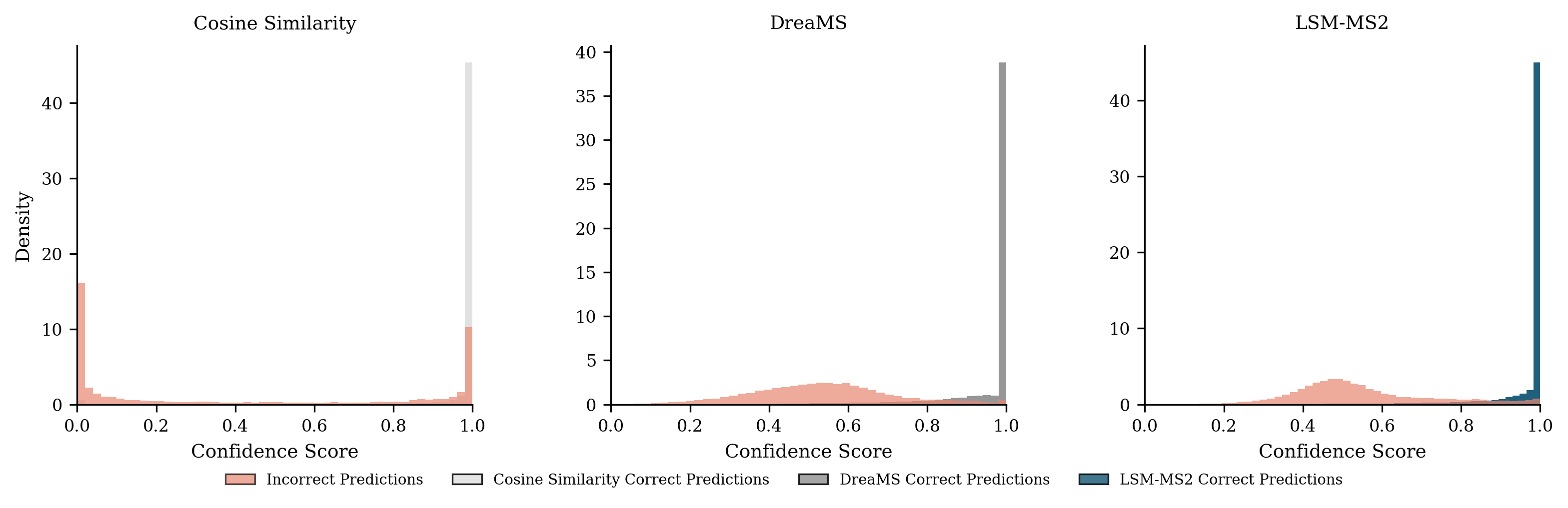
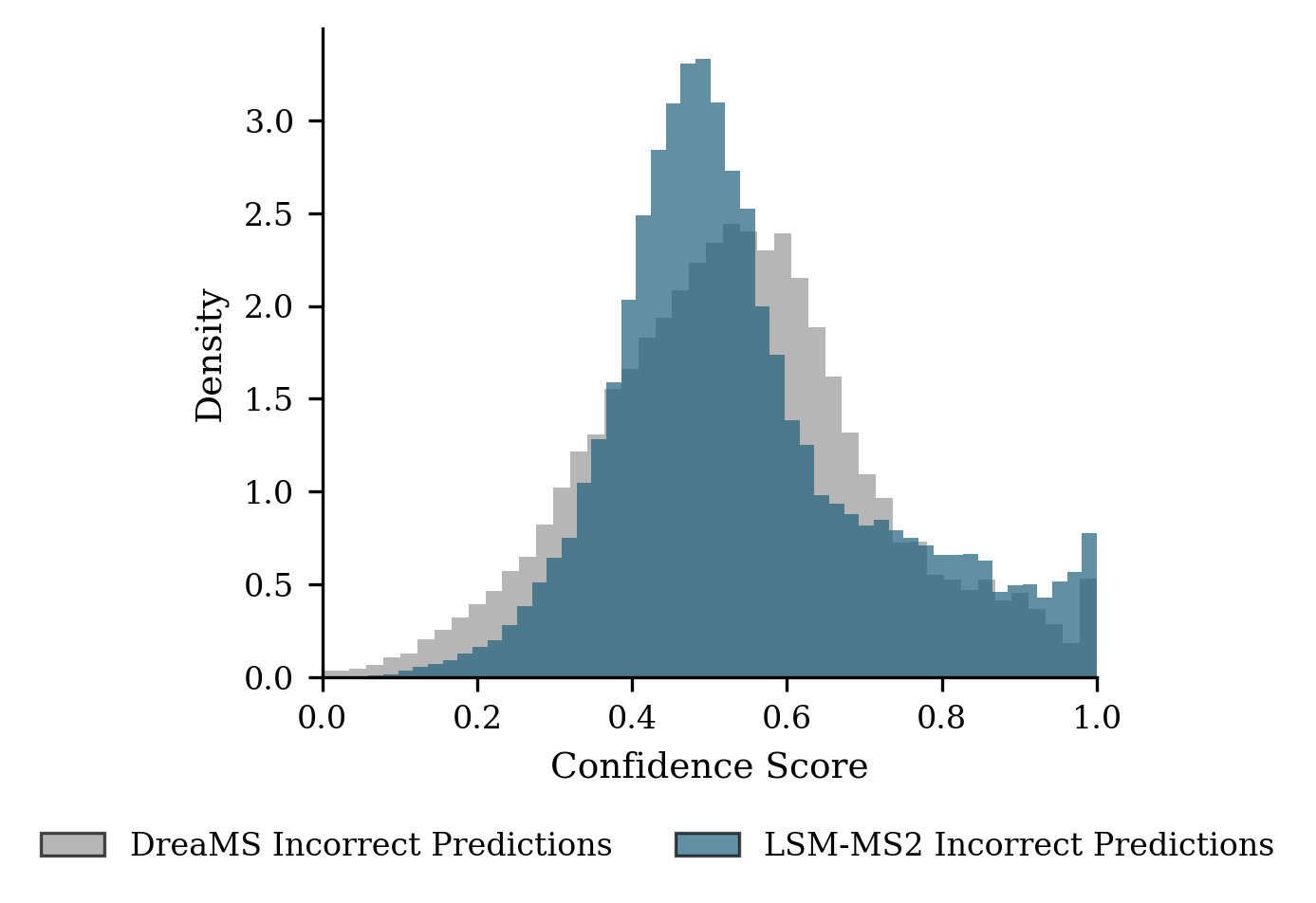
- On a major public benchmark (MassSpecGym), LSM-MS2 set a new state of the art, improving both accuracy and the quality of “near misses” (wrong answers that are still chemically close).
- It separates true matches from false matches more cleanly than other methods (better ROC/AUC), meaning it’s more reliable across scoring thresholds.
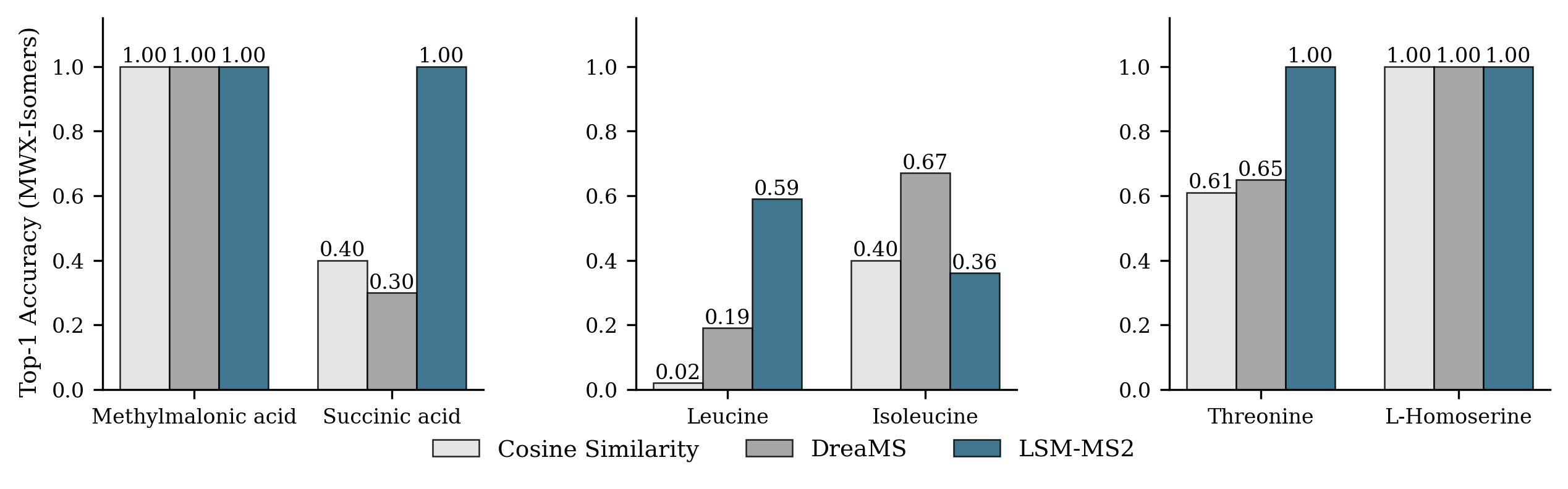
- Strong at telling apart very similar molecules (isomers):
- Isomers are like words made from the same letters arranged differently—same ingredients, different shapes and jobs in biology.
- LSM-MS2 improved isomer identification by about 30% over prior methods on a special isomer test set. It didn’t just get one isomer right in a pair—it balanced performance across both members, which is what really matters.
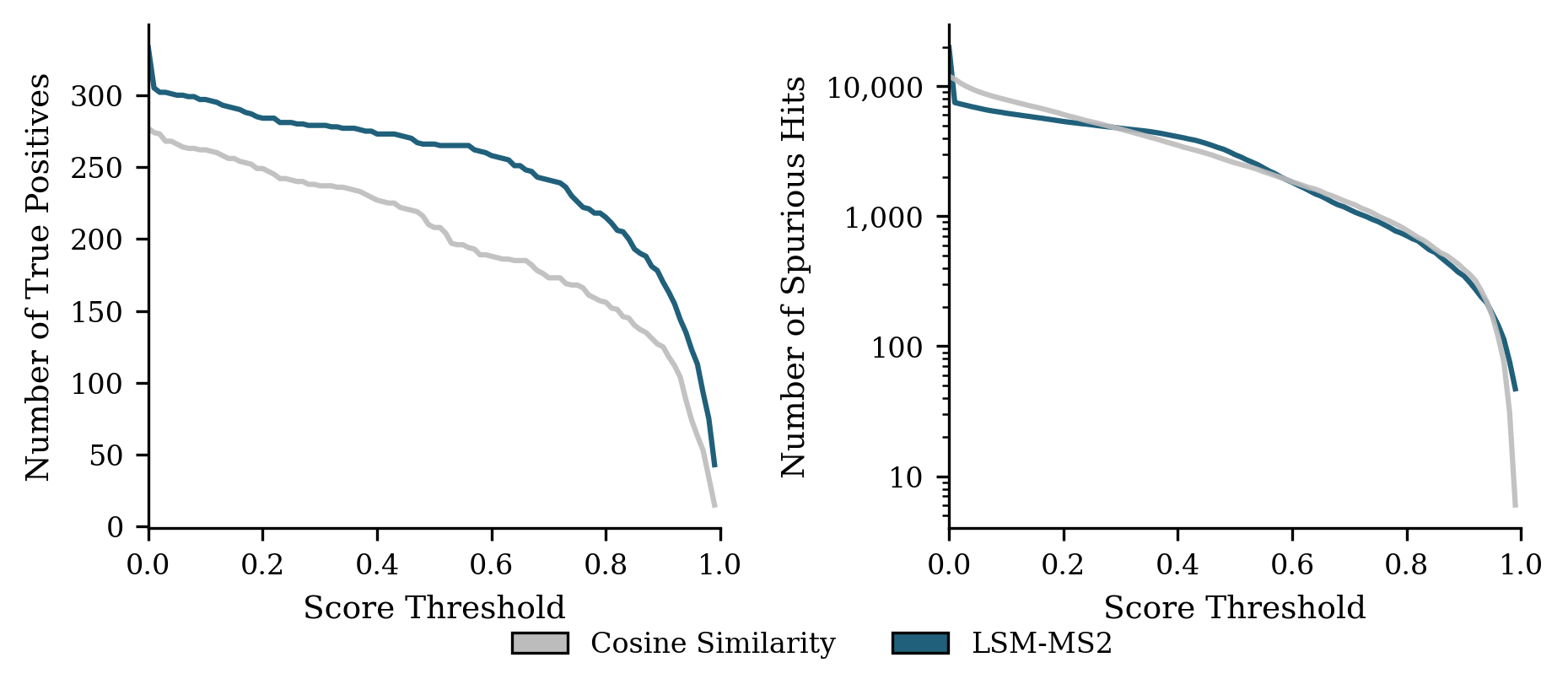
- Works well in complex, realistic samples and at low concentrations:
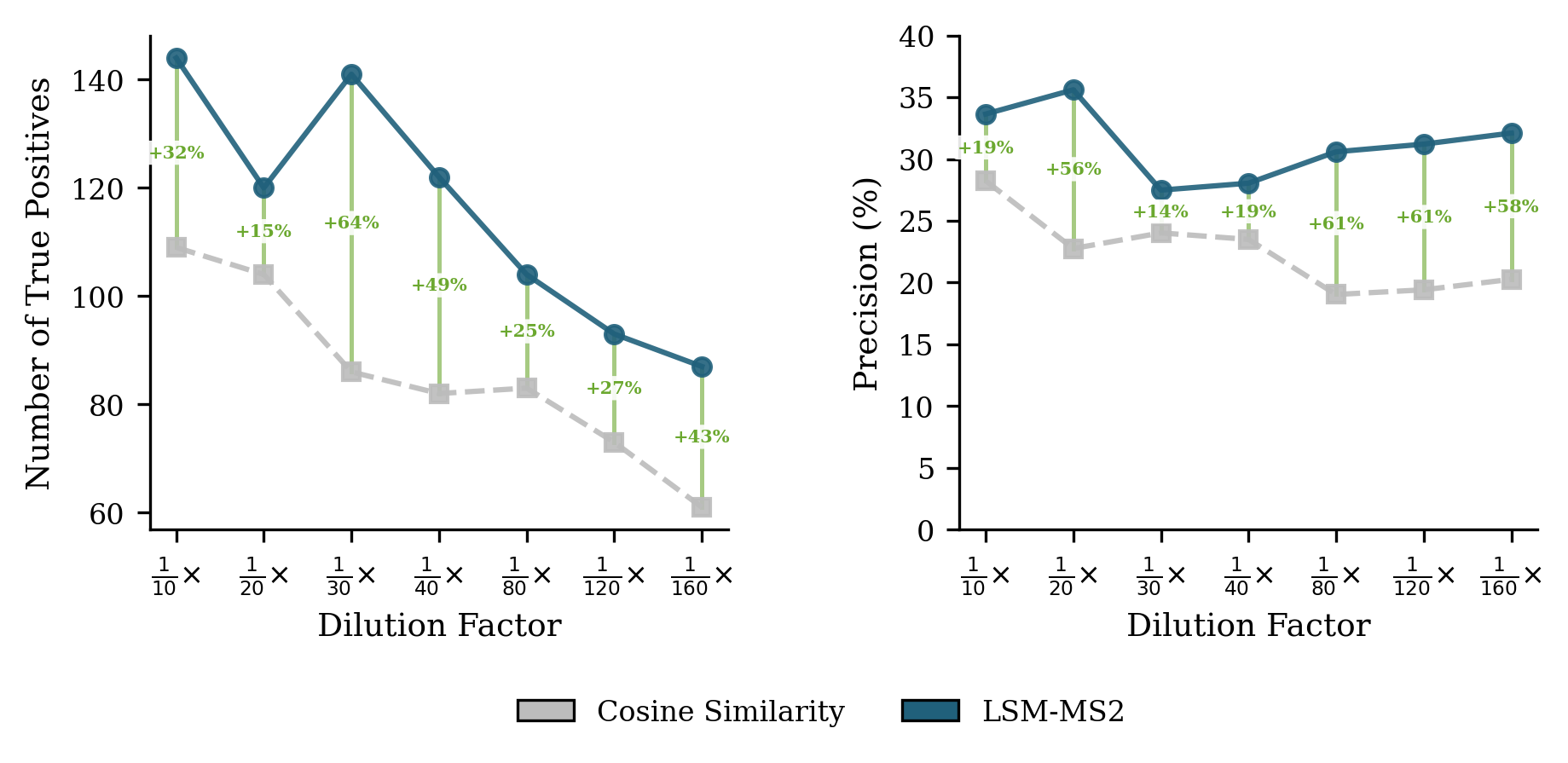
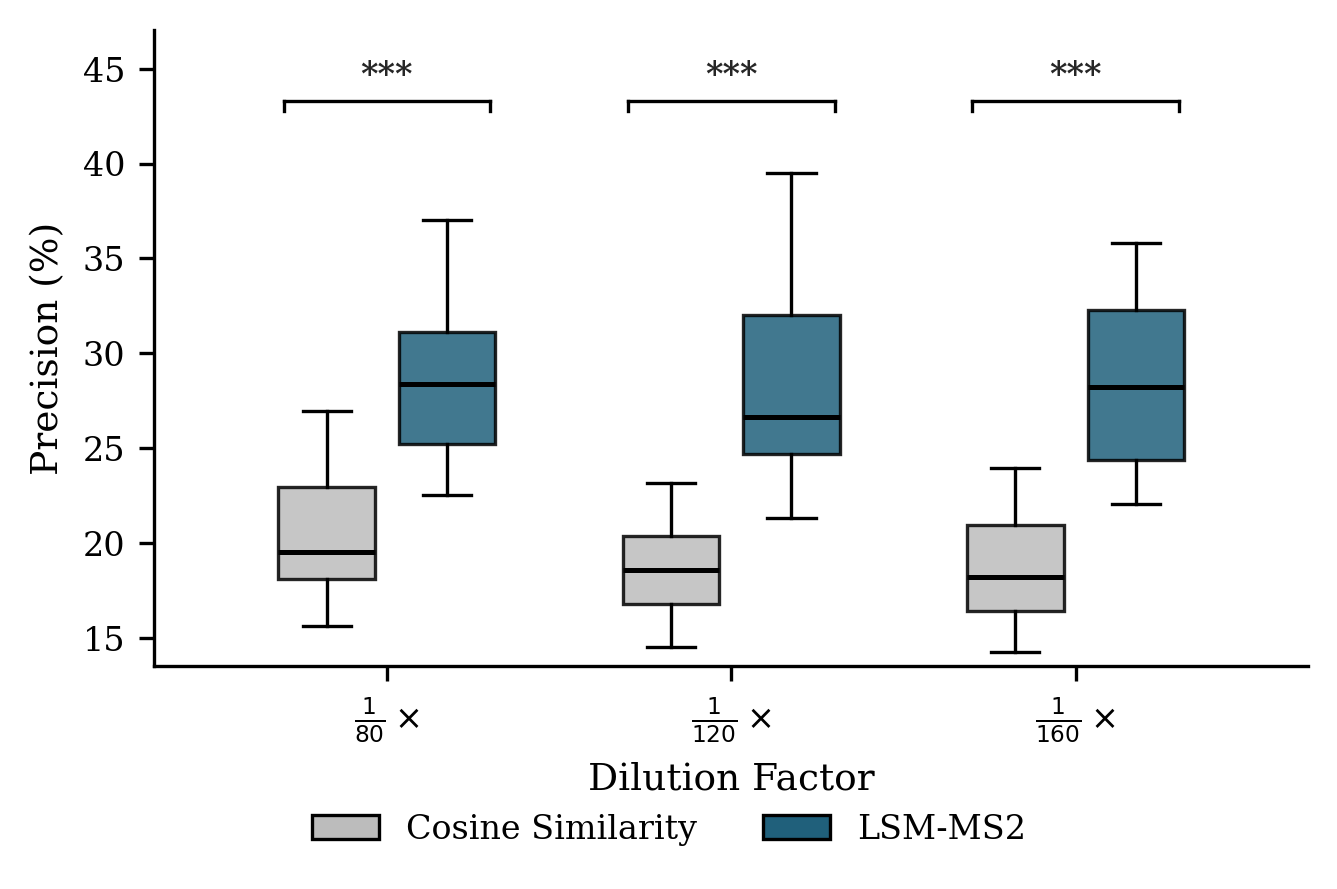
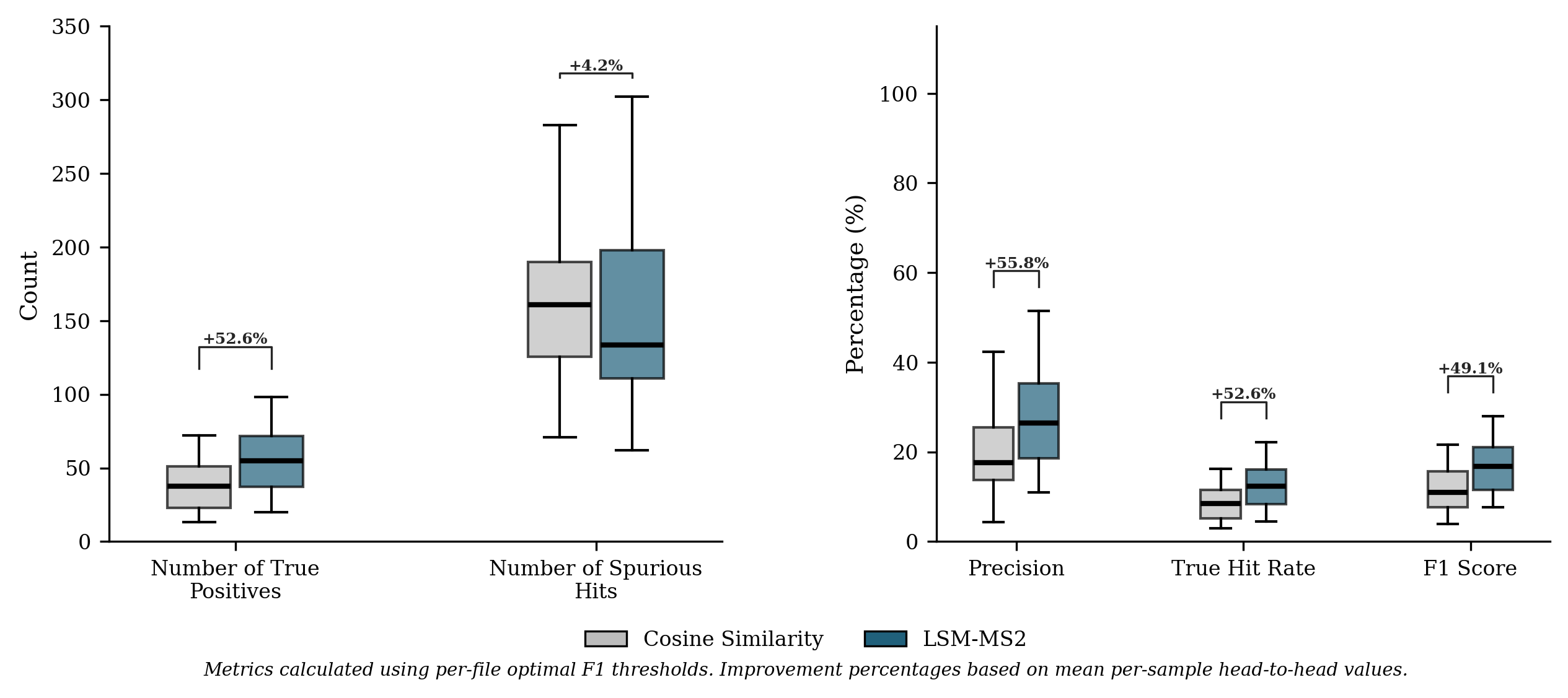
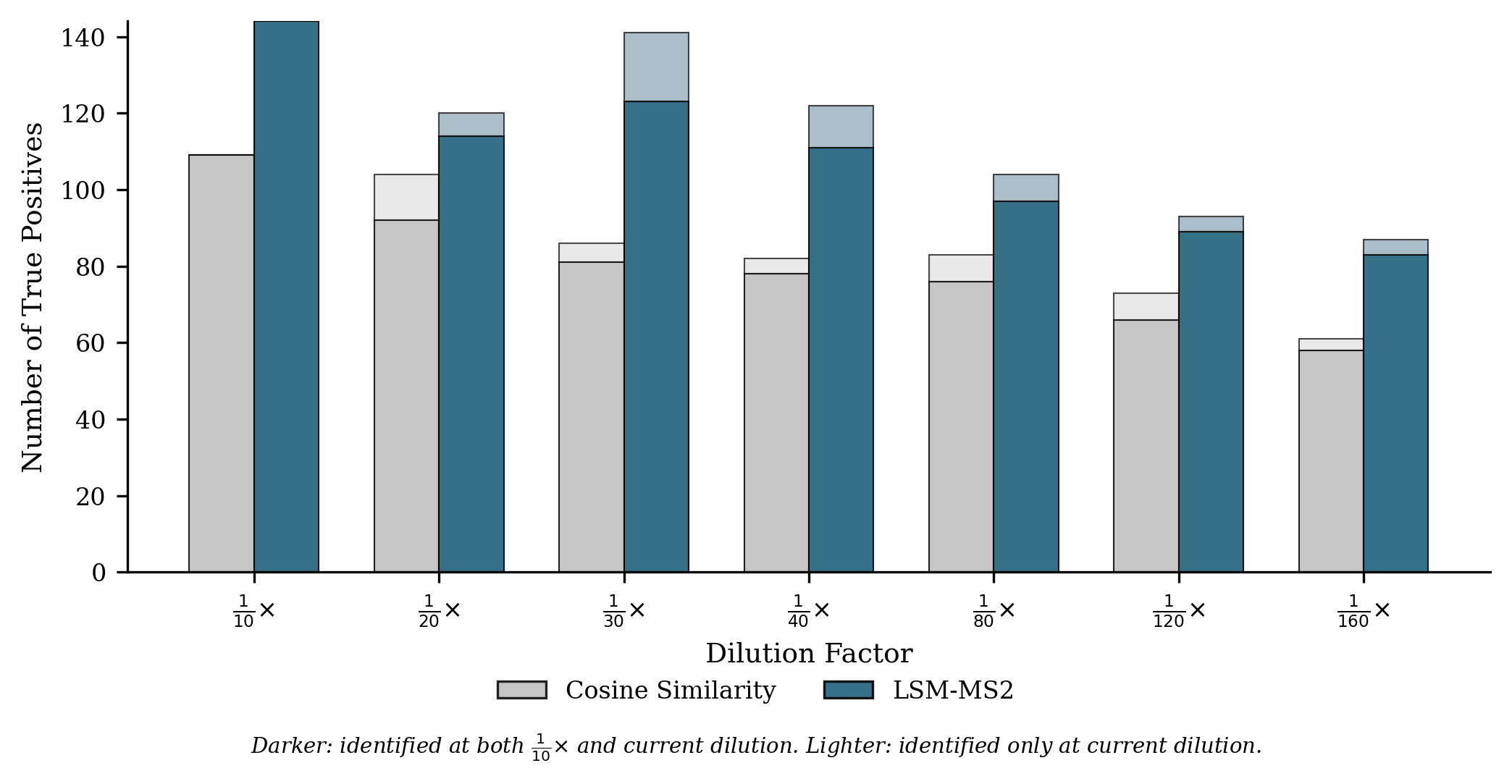
- In human plasma (NIST SRM 1950) across dilution series, LSM-MS2 found 42% more correct identifications at its best threshold than the classic cosine approach, with higher precision and robust performance even when signals were faint.
- Useful for biological insights—even without full identification:
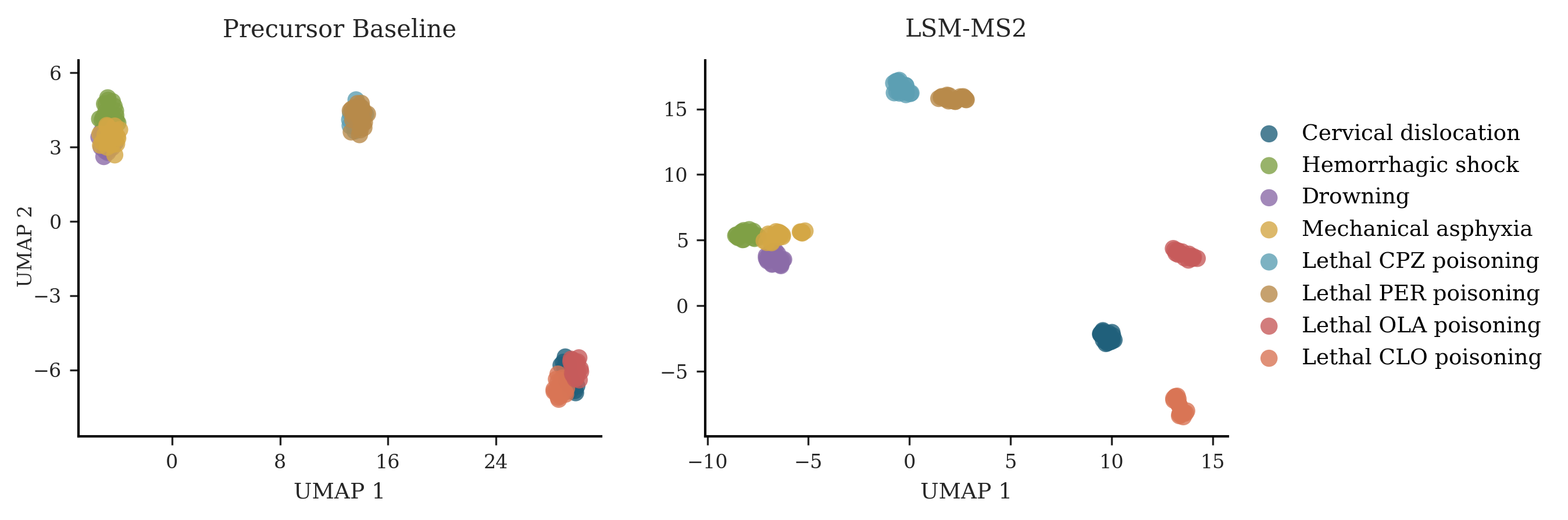
- Antipsychotic overdose classification: LSM-MS2’s embeddings naturally grouped causes of death more clearly than simple baselines, picking up meaningful metabolic patterns.
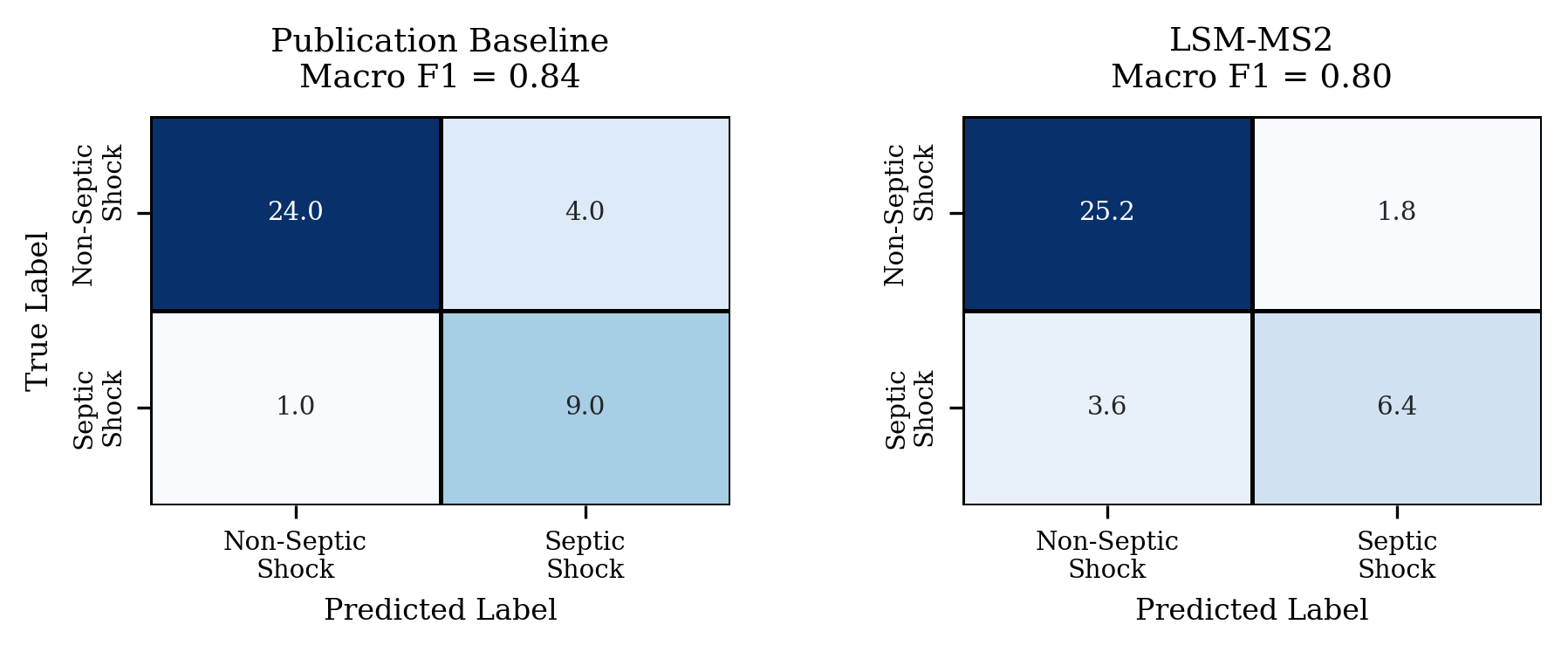
- Septic shock prediction: Using just the embeddings (no manual identification), it reached a macro F1 score of 0.80, close to the original study’s 0.84 that used a hand-picked set of identified metabolites—cutting analysis time dramatically.
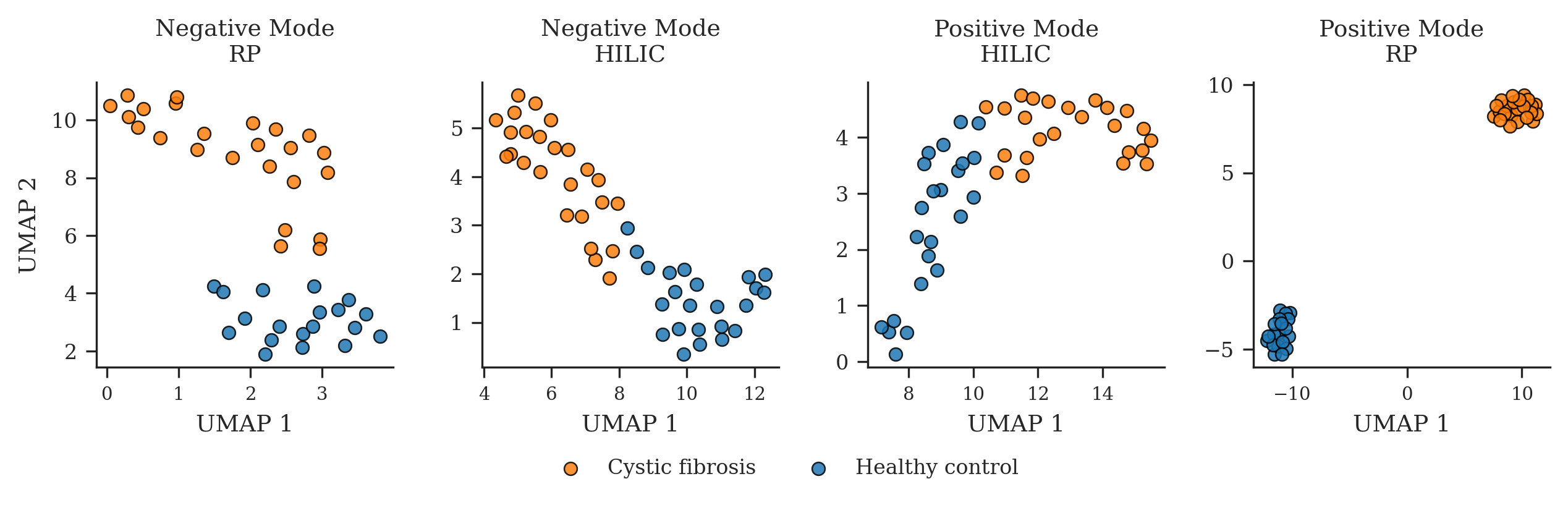
- Cystic fibrosis: The embeddings cleanly separated patient vs. control groups across multiple lab methods, and even suggested which lab setup (RP-positive mode) showed the clearest difference—helpful for designing future tests.
Why this matters: Most mass spectrometry data is “dark” (unlabeled). LSM-MS2 not only identifies more molecules, it also turns raw data into a meaningful map that can power quick biological conclusions, even when you don’t know the names of all the molecules yet.
What this could change (impact and future directions)
- Faster discoveries: Scientists can move from raw data to useful insights in minutes or hours, not days or weeks—especially helpful for small studies or early diagnostics.
- Better diagnostics and research: By recognizing disease patterns directly from spectra, labs can screen for conditions more efficiently, test hypotheses faster, and focus on what matters.
- Handling the unknown: Because the model learns a general “chemical map,” it can help with new or rare molecules that aren’t in libraries yet.
The authors plan to:
- Go beyond matching to generate molecule structures directly from spectra (so discoveries don’t depend on library coverage).
- Make the embeddings more interpretable, so we can see which spectral features drive differences between groups, helping translate patterns into clear biological mechanisms.
In short: LSM-MS2 is like a universal translator for mass spectrometry. It identifies more molecules, handles tricky lookalikes, stays strong when signals are weak, and helps scientists read the biological story hidden in raw spectra—quickly and reliably.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a single, consolidated list of what remains missing, uncertain, or unexplored in the paper. Each item is phrased to be specific and actionable for future research.
- Training transparency: the paper does not specify the exact training objective (e.g., InfoNCE, triplet loss), input representation (peak binning/tokenization, intensity scaling), model architecture details (parameter count, layers, positional encodings), optimization hyperparameters, or regularization strategies—hindering reproducibility and ablation studies.
- Dataset composition and diversity: the training corpus of “millions of spectra” lacks a breakdown by instrument type (Orbitrap, Q-TOF, IT), collision energies, fragmentation methods (HCD/CID/ETD), ionization modes, adducts, and chemical classes—making it unclear how well LSM-MS2 covers and generalizes across real-world acquisition variability.
- Train–test leakage risk: no statement confirms that spectra of benchmark analytes (MassSpecGym, MWX-Isomers, NIST) were excluded from pretraining; rigorous separation (e.g., by InChIKey or analyte family) is needed to rule out leakage and ensure unbiased evaluation.
- Reference library reproducibility: the 1.8M-spectrum library mixes public and internal sources; without public release or a fully reproducible build script, third parties cannot replicate retrieval results or quantify sensitivity to library composition.
- Baseline fairness and optimal use: DreaMS is evaluated via embedding cosine retrieval rather than its native scoring or pipeline; it remains unclear whether the comparison reflects DreaMS’s best-practice usage, and how results change with method-specific optimizations.
- Limited baselines for identification: comparisons omit strong hybrid tools (e.g., SIRIUS, MIST, CFM-ID, ESP) under matched library conditions; a broader baseline panel would clarify where LSM-MS2’s gains come from and when they persist.
- Structural similarity metric validation: the choice and parameterization of MCES (myopicMCES, threshold=15) is not justified or correlated to chemical or biological relevance; assessing multiple structure similarity metrics and their relationship to misidentification impact is needed.
- Isomer scope: evaluation excludes stereoisomers; actionable exploration of stereochemical discrimination (e.g., integrating ion mobility, retention time, chiral chromatography, or multi-energy MS2) is absent.
- Isomer-focused training: the paper demonstrates improved constitutional isomer separation without explicit contrastive supervision; open question: does targeted hard-negative mining on isomer groups further improve balanced discrimination across larger, more diverse isomer sets?
- Co-isolation and chimericity: the model’s behavior under cofragmentation, spectral leakage, and chimeric spectra is not directly evaluated; controlled tests with known chimeric mixtures are needed to quantify robustness.
- Score calibration and threshold stability: while ROC/AUC is reported for MassSpecGym, threshold transferability across instruments, methods, and matrices is not studied; calibration techniques (e.g., Platt scaling, conformal prediction) could improve reliability of decision thresholds.
- NIST ground truth approximation: treating “spurious hits” as false positives based on a subset of 443 known analytes is acknowledged as imperfect; acquiring orthogonal validation (e.g., authentic standards, retention time matching, MS3) would enable definitive precision/recall estimation.
- Fixed-threshold practicality: NIST analyses optimize F1 per dilution-specific threshold; real-world pipelines often require a single operating point—how does LSM-MS2 perform with fixed thresholds across dilutions, methods, and modes?
- AcquireX data omission: AcquireX spectra were collected but excluded from dilution series analysis; it remains unknown whether LSM-MS2’s gains hold under data-dependent enrichment strategies and iterative MS2 acquisition.
- Aggregation strategy for sample-level embeddings: the method for combining per-spectrum embeddings into a single sample representation is unspecified (e.g., mean/sum pooling, intensity weighting, attention, frequency normalization); ablations are needed to identify robust, bias-resistant aggregators.
- Batch effects and confounders: no assessment of batch effects (run order, instrument drift), matrix variability, or site/lab generalization is provided; cross-batch and cross-site validation is essential for translational reliability.
- Quantitative biological separation: CF analyses rely on UMAP visualization without supervised metrics (e.g., AUROC, F1, calibrated accuracy) or statistical tests; robustness across seeds and embedding hyperparameters, and evaluation against stronger baselines (feature detection + quantification + multivariate statistics), are missing.
- Biological baselines and controls: the MS1 “binned precursor” baseline is overly simplistic; comparisons should include conventional metabolomics workflows (feature finding, alignment, RT matching, normalization, identification, pathway-level modeling) and non-ML statistical pipelines.
- Downstream model details: for septic shock (macro F1=0.80), the classifier choice, hyperparameters, regularization, and feature preprocessing are not described; learning curves, calibration, and external validation against clinical cohorts are needed.
- Interpretability of embeddings: the paper proposes future work on interpretability but offers no current method to map spectral features (peaks, neutral losses, fragments) to sample-level differences; techniques like saliency, attention maps, spectral attribution, and substructure-level explanations should be developed and validated.
- OOD and novelty identification: performance when the correct analyte is absent from the library (true novelty) is not evaluated; open-set recognition, uncertainty quantification, and de novo structure inference benchmarks are necessary.
- Integration of orthogonal modalities: the model bypasses feature detection and RT usage; it remains unknown how incorporating retention time, ion mobility, MS1 isotopes, and chromatography metadata could improve identification and biological interpretation.
- Limits of low-concentration robustness: while dilution series results are promising, limits of detection/quantitation (LOD/LOQ), dynamic range, and sensitivity to matrix-specific noise are not quantified across sample types and instruments.
- Chemical space coverage and bias: the training and reference libraries may overrepresent certain classes (e.g., lipids, endogenous metabolites, pharmaceuticals); systematic evaluation of class-wise performance and rare class behavior is lacking.
- Statistical significance of SOTA gains: MassSpecGym improvements (e.g., Top-1 Acc. 0.739 vs 0.726) are not accompanied by statistical tests across multiple resamples or bootstraps; confidence intervals and significance analyses would strengthen claims.
- Scalability and compute: training/inference costs, memory footprint, latency, and throughput for large-scale experiments are not reported; practical deployment guidance (GPU/CPU requirements, batch sizing, model compression) is needed.
- Embedding dimensionality and scaling: the size and scaling properties of embeddings (e.g., concentration of measure, hubness) are unspecified; analyses of neighborhood structure, distance calibration, and cross-domain alignment would inform retrieval behavior.
- Safety and licensing: given proprietary components and patent claims, model/library/code availability, licensing terms, and constraints on academic/clinical use are unclear—limiting community validation and adoption.
- Cross-task generalization: beyond identification and three biological studies, broader tasks (pathway enrichment without IDs, mechanism inference, longitudinal trajectory analysis, treatment response prediction) are not explored; systematic benchmarks would demonstrate foundation-model versatility.
- Error analysis: the paper does not dissect common failure modes (e.g., adduct misassignment, in-source fragmentation, instrument-specific artifacts); targeted diagnostics could inform model and pipeline improvements.
- Instrument/method invariance: explicit tests of invariance to collision energy, fragmentation mode, resolution, and lab-specific tuning are missing; standardized cross-instrument transfer experiments should quantify robustness.
- Comparative use with hybrid tools: it remains unexplored whether LSM-MS2 embeddings can augment rule-based/generative tools (SIRIUS, MIST, CFM-ID) to improve identification and structural inference in a hybrid pipeline.
- Calibration to biological relevance: the relationship between structural similarity of misidentifications (MCES) and the biological interpretability of downstream models is not established; mapping chemical error types to biological decision impact is an open need.
Practical Applications
Immediate Applications
Below are actionable use cases that can be deployed now, leveraging the paper’s demonstrated performance gains in spectral identification, isomer discrimination, robustness at low concentrations, and annotation-free biological interpretation via embeddings.
- Healthcare — Emergency triage for septic shock risk prediction
- Use case: Generate sample-level embeddings from admission serum LC–MS/MS, train a simple classifier (e.g., logistic regression) to predict early onset of septic shock, achieving macro F1 ≈ 0.80 without manual identification, enabling hour-scale turnaround for model creation and deployment.
- Tools/workflows: LSM-MS2 embedding extractor → per-file aggregation → classifier training → threshold calibration (AUC-guided) → clinical reporting.
- Dependencies/assumptions: CLIA/FDA validation, site-specific calibration, standardization of LC–MS acquisition, data governance and privacy, model monitoring for drift.
- Healthcare — Method selection assistant for disease-specific assays (e.g., cystic fibrosis)
- Use case: Use unsupervised clustering on sample-level embeddings to choose optimal LC/ionization mode that maximizes cohort separation (e.g., RP positive mode for CF), accelerating assay development.
- Tools/workflows: LSM-MS2 embeddings → UMAP/TSNE visualization → cohort separability metrics → method recommendation.
- Dependencies/assumptions: Access to representative pilot data, consistent instrument settings, awareness that separability may be biomarker or mode specific.
- Forensic toxicology — Cause-of-death classification for antipsychotic overdoses
- Use case: Triage and classify fatalities into CPZ/PER/OLA/CLO vs non-drug controls using LSM-MS2 embeddings, improving resolution vs precursor-only baselines and reducing manual identification burden.
- Tools/workflows: Embedding-based clustering and classification, chain-of-custody compliant pipelines, score thresholding with ROC-guided calibration.
- Dependencies/assumptions: Jurisdiction-specific validation, instrument cross-compatibility, standardized sample prep and acquisition, legal admissibility.
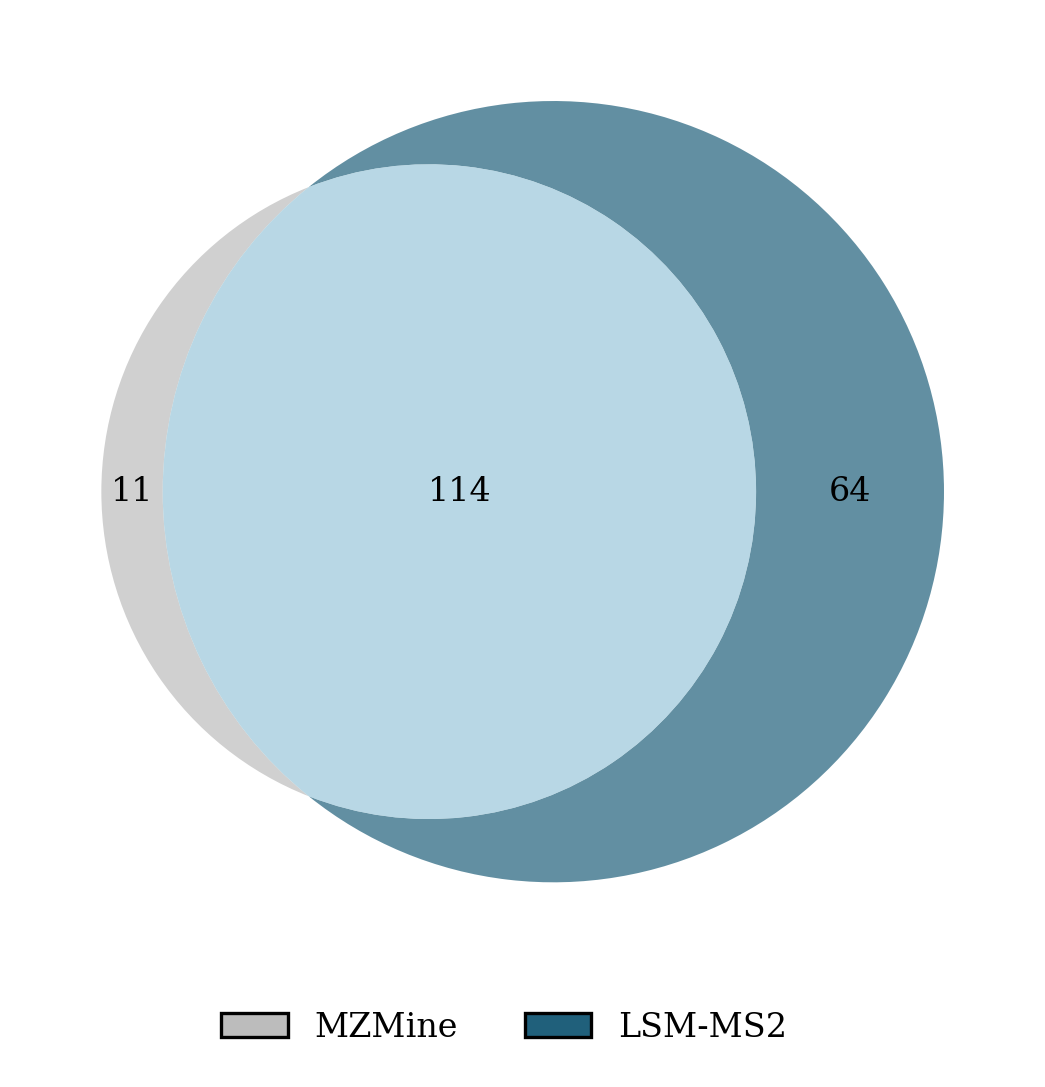
- Metabolomics core labs — Higher-yield spectral identification
- Use case: Integrate LSM-MS2 into retrieval pipelines (e.g., MZmine, GNPS) to increase Top-1 accuracy (state-of-the-art on MassSpecGym), retrieve analytes more chemically similar when wrong (lower MCES), and improve separation of true vs false hits (AUC ≈ 0.972).
- Tools/products: LSM-MS2 library search engine, MZmine/GNPS plugin, score threshold optimizer, batch QC dashboard.
- Dependencies/assumptions: Reference library coverage limits maximum achievable accuracy; careful threshold selection per dataset; ongoing curation and instrument calibration.
- Pharma/biotech — Isomer-resolver for analytical QC and pharmacology
- Use case: Apply improved isomeric discrimination (≈30% more correct analyte calls; balanced separation across isomer pairs like leucine/isoleucine) in bioanalysis, metabolite ID, and target engagement studies.
- Tools/workflows: Embedding-based retrieval with isomer group-level reporting, differential scoring for closely related structures, integration with SIRIUS/MIST-CF for formula constraints.
- Dependencies/assumptions: Primarily constitutional isomers; stereoisomer resolution still limited with MS2 alone; method orthogonality (e.g., ion mobility) may be needed.
- Complex matrices and low-abundance analytes — Increased true positives without added noise
- Sector: Healthcare, environmental testing, food safety.
- Use case: In plasma-like matrices, LSM-MS2 retrieves ≈42% more true positives than cosine with ≈33% higher precision at optimal F1, maintaining precision across dilutions; applicable to trace detection in QC and surveillance.
- Tools/workflows: Threshold optimization per dilution/matrix, precision-focused reporting, spurious-hit auditing.
- Dependencies/assumptions: Matrix-specific validation; spurious-hit definitions must be aligned to ground-truth sets.
- Bioprocess and manufacturing — Run-to-run QC via sample-level embeddings
- Use case: Monitor batch consistency and detect shifts using embedding similarity, enabling rapid flagging of process deviations without full identification.
- Tools/workflows: Embedding-based statistical process control, drift dashboards, anomaly detectors.
- Dependencies/assumptions: Stable acquisition protocols, LIMS integration, periodic recalibration.
- Academia and education — Rapid hypothesis generation on small datasets
- Use case: Replace multi-week identification workflows with embedding-based clustering and modeling to discern phenotypic differences (e.g., disease vs control) in small sample studies (<100).
- Tools/workflows: Open-source embedding pipelines, UMAP viewers, simple classifiers, reproducible seeds for stratified splits.
- Dependencies/assumptions: Access to raw MS/MS files, consistent metadata, training in minimal ML.
- Software and data infrastructure — Embedding-as-a-service
- Sector: Software/Cloud.
- Use case: Offer a cloud API or on-prem extractor to produce spectrum and sample embeddings, plus libraries for retrieval/scoring and method selection.
- Tools/products: LSM-MS2 SDKs, containerized services, GNPS/MZmine connectors.
- Dependencies/assumptions: Licensing, compute costs (GPU for batch inference), version control, data standardization (mzML/mgf).
Long-Term Applications
These use cases require further research, scaling, validation, or development (e.g., generative structure inference, interpretability, regulatory approval, or instrument firmware integration).
- Generative “structure-from-spectrum” engines
- Sector: Software, pharma, biotech.
- Use case: Infer molecular structures de novo from spectra to reduce library dependence and discover novel compounds; integrate with knowledge graphs for pathway mapping.
- Tools/products: Generative LSM-MS2, confidence scoring, semi-supervised workflows, human-in-the-loop curation.
- Dependencies/assumptions: Robust calibration of generative confidence, evaluation benchmarks beyond retrieval (CASMI-like), large-scale validation.
- Real-time on-instrument decisioning and robotics
- Sector: Robotics/instrumentation.
- Use case: Adaptive DDA/DIA guided by embeddings—on-the-fly selection of collision energies/precursor targets and closed-loop acquisition optimizing coverage and isomer separation.
- Tools/products: Firmware APIs for latency-constrained inference, acquisition planners, reinforcement learning controllers.
- Dependencies/assumptions: Vendor collaboration, real-time compute constraints, stringent QA, safety and reliability testing.
- Clinically approved diagnostics from embeddings (multi-center)
- Sector: Healthcare.
- Use case: Embedding-based assays for septic shock triage, CF monitoring, and other metabolomic diseases; EHR-integrated risk scores with reimbursement.
- Tools/products: Validated clinical decision support systems, assay kits, quality-controlled pipelines, post-market surveillance.
- Dependencies/assumptions: Regulatory approvals (FDA/EMA), prospective trials, standardized protocols, bias/robustness analysis across sites.
- Environmental and food supply chain “Spectral Sentinel”
- Sector: Policy/public health, agriculture, food safety.
- Use case: Networked LC–MS surveillance using embeddings to detect emerging contaminants at low abundance; rolling baselines for regional matrices.
- Tools/products: Cloud dashboards, anomaly detection, alerting systems, cross-lab harmonization.
- Dependencies/assumptions: Data-sharing agreements, standardized acquisition, governance frameworks for action thresholds.
- Drug discovery — Phenotypic screening and target deconvolution
- Sector: Pharma/biotech.
- Use case: Use embeddings to cluster phenotypic responses, prioritize hits, and link spectral shifts to mechanisms/pathways via integrated interpretability and curated knowledge bases.
- Tools/products: “Mechanism mapper” linking embedding features to pathways, integration with transcriptomics/proteomics, multimodal foundation models.
- Dependencies/assumptions: Improved interpretability linking spectral features to biology, multimodal data alignment, expert curation.
- Enhanced isomer and stereoisomer resolution via multimodal integration
- Sector: Analytical chemistry, instrumentation.
- Use case: Combine MS2 embeddings with ion mobility, retention time modeling, and orthogonal chemistries to discriminate stereoisomers and complex isomer families.
- Tools/products: Multimodal embedding fusion, retention-index predictors, cross-technique calibration.
- Dependencies/assumptions: Access to orthogonal modalities, unified training datasets, instrument synchronization.
- Standards, benchmarks, and certification for MS foundation models
- Sector: Policy/standards, academia.
- Use case: Establish community benchmarks for interpretability and clinical validity; certify model versions for specific use contexts (clinical, QC, forensics).
- Tools/products: Extended MassSpecGym-like suites (biological endpoints, confidence metrics), model cards, audit trails.
- Dependencies/assumptions: Community consensus, reproducibility requirements, regulatory collaboration.
- Consumer and preventive health metabolomics (longer horizon)
- Sector: Consumer health.
- Use case: Embedding-based wellness profiling and longitudinal monitoring with standardized sample kits and cloud analytics to flag metabolic deviations.
- Tools/products: At-home sampling kits, secure mobile apps, personalized dashboards.
- Dependencies/assumptions: Simplified, robust sample collection, strong privacy protections, medical oversight, clear benefit-risk frameworks.
- Workforce and training impacts
- Sector: Education/industry.
- Use case: Upskilling programs for embedding-based LC–MS analytics; curricula emphasizing foundation models, thresholding, and interpretability.
- Tools/products: Certification courses, sandbox datasets, teaching modules.
- Dependencies/assumptions: Broad access to tools/datasets, institutional buy-in, ongoing model updates.
Cross-cutting assumptions and dependencies to consider
- Data: Quality-controlled MS/MS acquisition, consistent collision energies and instrument tuning, curated reference libraries; small-task datasets may require careful class balance and stratification.
- Model scope: Identification accuracy remains library-limited; stereoisomers are challenging for MS2 alone; embeddings provide phenotypic separations but may need interpretability for mechanism.
- Operationalization: Score threshold calibration is method- and dataset-specific; instrument and firmware compatibility; compute requirements for high-throughput inference; LIMS/EHR integration.
- Governance: Regulatory approvals for clinical decision support; privacy/compliance for health and surveillance applications; reproducibility, auditability, and bias monitoring across sites.
Glossary
- AcquireX: Thermo Fisher’s iterative data acquisition workflow that enhances coverage by repeatedly targeting unobserved or low-quality features. "AcquireX data were excluded for this analysis."
- Analyte: A distinct chemical compound measured or identified in a mass spectrometry experiment. "Our reference library comprises 1.8 million high-quality spectra corresponding to 99 thousand unique analytes."
- AUC: Area Under the ROC Curve; a scalar metric summarizing the trade-off between true positive rate and false positive rate across thresholds. "The resulting area under the curve (AUC) is 0.950, 0.965, and 0.972 for Cosine Similarity, DreaMS, and LSM-MS2, respectively"
- CASMI: Critical Assessment of Small Molecule Identification; a community benchmark for evaluating automated small-molecule identification methods. "public benchmarks for systematic comparison, such as MassSpecGym and CASMI."
- Collision energy: The energy applied during MS/MS that controls how precursor ions fragment, shaping the resulting spectrum. "experimental variability (e.g. collision energy and instrument type);"
- Constitutional isomer: Compounds with the same molecular formula but different connectivity of atoms. "Only constitutional isomers were included, since stereoisomers cannot be reliably distinguished using MS data."
- DreaMS: A self-supervised foundation model for MS/MS spectra used for spectral identification and representation learning. "DreaMS, in contrast, focuses on small molecules and was trained on a chemical space similar to ours; yet it falls short of LSM-MS2 in spectral retrieval performance and does not demonstrate downstream biological interpretation."
- Fragment ion: An ion produced by the breakup of a precursor ion during tandem mass spectrometry. "with precursor and fragment ions measured at single–part-per-million (ppm) mass accuracy"
- Foundation model: A large-scale pretrained model that learns generalizable representations applicable to many downstream tasks with minimal fine-tuning. "Foundation models offer a solution through learning rich, generalizable representations from large datasets, enabling multiple downstream tasks with minimal fine-tuning"
- GNPS: Global Natural Products Social Molecular Networking; a platform for sharing and analyzing mass spectrometry data. "Compound Discover, Global Natural Products Social Molecular Networking (GNPS), and MZmine."
- HILIC: Hydrophilic Interaction Liquid Chromatography; a separation technique optimized for polar compounds. "We collected 84 samples with RP and HILIC methods in positive and negative modes"
- InChIKey: A hashed textual identifier derived from an InChI that uniquely represents a chemical structure. "We also identified 272 spectra where the provided InChIKeys did not match those generated from the associated isomeric SMILES"
- Ionization mode: The polarity setting (positive or negative) under which ions are generated and detected in mass spectrometry. "in positive and negative ionization modes."
- Isomeric discrimination: The ability to differentiate and correctly identify members of an isomer group from MS/MS data. "Genuine isomeric discrimination requires that all unique members of an isomer group are correctly identified; otherwise, the model has not truly learned to separate the isomers."
- LC–MS: Liquid Chromatography–Mass Spectrometry; an analytical technique coupling chromatographic separation with mass analysis. "Mass spectrometry coupled with liquid chromatography (LC–MS) offers a dense view of the molecular state of biological systems."
- MassSpecGym: A comprehensive benchmark dataset for evaluating MS/MS identification and discovery models. "MassSpecGym: The most comprehensive public benchmark for MS/MS data, containing 231 thousand high-quality spectra spanning 29 thousand analytes."
- Mass-to-charge ratio (m/z): The fundamental measurement axis in mass spectrometry representing ion mass divided by its charge. "Each sample is represented by a 0–1000~m/z binned vector"
- Maximum Common Edge Subgraph (MCES) Distance: A graph-based measure of structural dissimilarity between two molecular graphs computed via their largest shared edge subgraph. "Top-K Maximum Common Edge Subgraph (MCES) Distance: Measures structural similarity between predicted and ground-truth molecular graphs as an edit distance"
- MS1: The first stage of mass spectrometry that measures intact precursor ions without fragmentation. "MS1 only (precursor) baseline embedding"
- MS2: Tandem mass spectrometry stage where selected precursor ions are fragmented to produce detailed structural information. "Machine learning (ML) on tandem mass spectrometry (MS/MS or MS2) provides a framework to analyze this high-dimensional and unstructured data"
- MZmine: An open-source software platform for processing and analyzing mass spectrometry data. "MZmine hyperparameters were selected to maximize algorithmic comparability between methods rather than to individually optimize either approach."
- myopicMCES: A specific implementation/tool used to compute MCES distance under predefined constraints. "Computed using myopicMCES (threshold=15)."
- NIST SRM 1950: A standardized human plasma reference material used for benchmarking analytical performance. "NIST SRM 1950 human plasma dilution series used to evaluate performance in a biologically complex medium"
- Precursor ion: The selected ion that undergoes fragmentation in MS/MS to generate a spectrum of fragment ions. "with precursor and fragment ions measured at single–part-per-million (ppm) mass accuracy"
- Reversed-phase (RP): A chromatography mode featuring a nonpolar stationary phase, commonly used for separating less polar compounds. "Samples were profiled using both reversed-phase (RP) and hydrophilic interaction (HILIC) liquid chromatography"
- ROC curve: Receiver Operating Characteristic curve; plots true positive rate against false positive rate across score thresholds. "using ROC curves, which plot the true positive rate (TPR) against the false positive rate (FPR) across all scoring thresholds."
- Spectral leakage: Undesired signal spillover or contamination that obscures true spectral features. "spectral leakage; and noisy peaks that obscure informative signals"
- Spectral library: A curated collection of reference mass spectra for known compounds used to identify experimental spectra. "public spectral libraries"
- Spectral embedding: A learned vector representation of mass spectra capturing chemical semantics for downstream tasks. "rich spectral embeddings that enable direct biological interpretation from minimal downstream data"
- Spectral identification: The process of matching an experimental spectrum to spectra of known compounds to determine its identity. "Spectral identification measures how well a model matches an experimental (\"test\") spectrum to those from known reference compounds."
- SMILES (isomeric): A textual representation of molecular structures; the isomeric variant encodes stereochemistry and isotopic information. "associated isomeric SMILES"
- ppm (part-per-million) mass accuracy: A precision measure indicating how closely measured masses match true values, expressed per million units. "single–part-per-million (ppm) mass accuracy"
- UMAP: Uniform Manifold Approximation and Projection; a nonlinear dimensionality reduction method for visualization. "Unsupervised UMAP projections of study samples colored by mortality cause."
Collections
Sign up for free to add this paper to one or more collections.

