- The paper introduces a formal goodness-of-fit procedure that models sampling-induced distortions in observed degree distributions using a transformation matrix.
- It leverages GMM for parameter estimation and a custom parametric bootstrap to control type I error and enhance test power for different network models.
- Application to yeast protein interaction networks decisively rejects the Poisson model, supporting the prevalence of heavy-tailed, scale-free characteristics.
Goodness-of-Fit Testing for Degree Distributions from Induced Subgraphs
This work addresses the statistical problem of testing whether the degree distribution of a population graph G conforms to a specified family (e.g., Poisson, power law), when only an induced subgraph sample G′ is observed. Unlike i.i.d. sampling of vertex degrees, induced subgraph sampling systematically distorts the observed degree sequence because larger-degree vertices are more likely to have connections present in the sample, which complicates standard goodness-of-fit (GoF) testing and parametric estimation procedures.
The motivating application is in network science, particularly the inference of biological networks such as protein interaction networks (PINs), which are frequently measured through protocols (e.g., yeast two-hybrid assays) that result in induced subgraph data. Understanding the true degree distribution of such networks has significant implications for identifying essential proteins and designing interventions.
Statistical Methodology
The central contribution is a formal GoF testing procedure for degree distribution families on induced subgraph data, combining several key components:
1. Distortion Modeling:
The expectation of the sampled degree distribution PG′ given the population degree distribution PG is formalized as E[PG′]=XPG, where X encodes the sampling-induced transformation (with hypergeometric or binomial structure depending on sampling design). Direct inversion is ill-posed due to high dimensionality and the ill-conditioning of X.
2. GMM Parameter Estimation:
Parameters θ of the hypothesized family F0(θ) are estimated via Generalized Method of Moments, minimizing a quadratic loss between the observed degree frequencies and the image of the hypothesized distribution under the sampling operator X, using an estimated optimal covariance weighting for efficiency and convexity.
3. Goodness-of-Fit Statistic:
A Kolmogorov-Smirnov type statistic is constructed using the empirical CDF from G′ and the CDF implied by F0(θ^), propagated through X. The difficulty lies in characterizing the null distribution of the test statistic since the hypothesized family is only implicitly defined through θ estimated from the same data.
4. Graphical Parametric Bootstrap:
The principal innovation is the introduction of a parametric bootstrap customized for induced subgraph sampling. For each bootstrap iteration, a random (possibly multi-)graph matching the estimated degree distribution is synthesized (via the configuration model), and an induced subgraph is resampled under the same sampling protocol as the data. Critical values for the GoF statistic are thus empirically simulated, appropriately accounting for both estimation and sampling-induced distortions.
Simulation Study
A comprehensive Monte Carlo study investigates the type I error control (size) and power of the proposed procedure under common network degree distributions:
- Null families: Poisson (Erdős–Rényi) and scale-free (power law) with a range of parameters.
- Sampling design: Simple random sampling of vertices at varying rates.
- Key results: When the true underlying distribution matches the null hypothesis, type I error is held closely to the nominal level (≈5%), demonstrating robust control. Power increases monotonically with sample size and is particularly strong for larger differences between Poisson and scale-free parameters.
Notably, in regimes where degree distributions nearly overlap (e.g., low Poisson rate vs. extremely heavy-tailed power-law), the procedure—like any degree-based hypothesis test—exhibits lower discriminability.
Application: Protein Interaction Networks
The framework is applied to multiple high-quality Saccharomyces cerevisiae PINs published by Yu et al., incorporating explicit modeling of high false negative rates inherent in Y2H assay data. Essential technical steps include:
- Modeling edges in the subgraph as observed independently with success rate r (true negative rate), and adapting the X matrix accordingly (Poisson–binomial convolution).
- Hypothesis tests are performed for Poisson, exponential, and scale-free degree distributions across a suite of PIN datasets and for several assumed false negative rates (70–90%).
The empirical results are decisive:
- Poisson (Erdős–Rényi) family hypotheses are unambiguously rejected (p-value ≈ 0) for all datasets and all plausible false negative rates.
- The scale-free hypothesis is generally retained for most datasets at moderate to high false negative rates, but not universally—emphasizing the nuances of network growth and measurement errors in real interactome data.
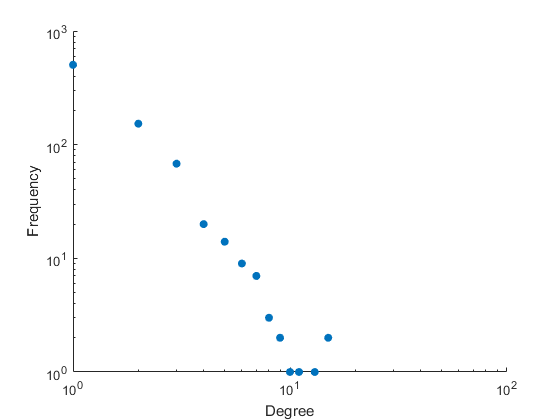
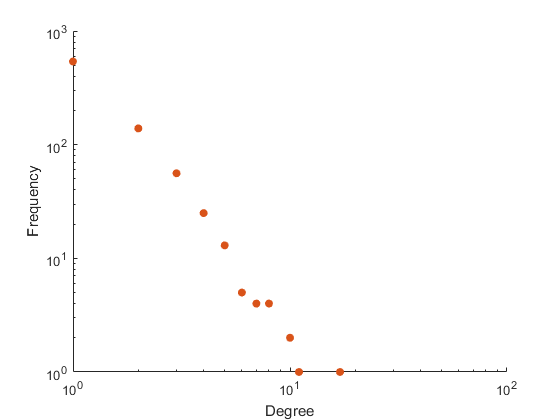
Figure 2: Empirical degree distribution (log-log scale) for the Ito-core yeast PIN, illustrating the fit (or lack thereof) to scale-free and Poisson alternatives.
Theoretical Implications and Limitations
This work generalizes GoF testing for network degree distributions to the induced subgraph sampling context, extending classical Lilliefors testing to a combinatorial domain where complex, parameter-dependent distortions are present. The framework is compatible with any parametric degree distribution and, crucially, is directly extensible to other node-sampled designs where the sampling operator X is linear and population-invariant.
The main practical limitation is computational: the generation of large random graphs for the bootstrap and the storage of adjacency matrices is prohibitive for massive graphs (e.g., networks with millions of vertices). There is an open opportunity for scalable or graph-free analogs.
Future Directions
Several research directions are immediate:
- Algorithmic scalability: Developing alternatives to explicit population-size graph generation for the bootstrap in massive graphs.
- Broader sampling designs: Extending to snowball, traceroute, or egocentric designs by suitably adapting the X operator.
- Refined models of measurement error: Incorporating correlated errors, context-specific biases, and adaptive estimation strategies.
- Inference for other graph properties: Extending the paradigm to motifs, clustering coefficients, or community structure statistics.
Conclusion
This work provides a statistically principled and computationally feasible procedure for testing degree distributional hypotheses for large graphs when only induced subgraphs are accessible. The proposed graphical bootstrap method effectively adapts the GoF paradigm to the constraints of practical sampling, as validated by theoretical analysis, simulation, and application to high-impact biological networks. Empirically, the rejection of the Poisson family for yeast PINs provides further evidence against Erdős–Rényi random graph models for biological systems, reinforcing the importance of heavy-tailed mechanisms in cellular interaction networks.
Reference:
"A Goodness-of-Fit Test for Sampled Subgraphs" (1710.04801)