Pearl: A Foundation Model for Placing Every Atom in the Right Location
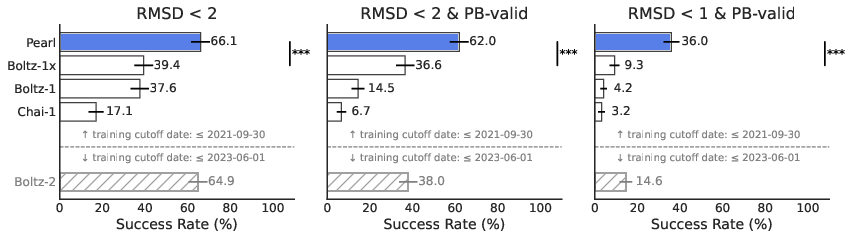
Abstract: Accurately predicting the three-dimensional structures of protein-ligand complexes remains a fundamental challenge in computational drug discovery that limits the pace and success of therapeutic design. Deep learning methods have recently shown strong potential as structural prediction tools, achieving promising accuracy across diverse biomolecular systems. However, their performance and utility are constrained by scarce experimental data, inefficient architectures, physically invalid poses, and the limited ability to exploit auxiliary information available at inference. To address these issues, we introduce Pearl (Placing Every Atom in the Right Location), a foundation model for protein-ligand cofolding at scale. Pearl addresses these challenges with three key innovations: (1) training recipes that include large-scale synthetic data to overcome data scarcity; (2) architectures that incorporate an SO(3)-equivariant diffusion module to inherently respect 3D rotational symmetries, improving generalization and sample efficiency, and (3) controllable inference, including a generalized multi-chain templating system supporting both protein and non-polymeric components as well as dual unconditional/conditional modes. Pearl establishes a new state-of-the-art performance in protein-ligand cofolding. On the key metric of generating accurate (RMSD < 2 \r{A}) and physically valid poses, Pearl surpasses AlphaFold 3 and other open source baselines on the public Runs N' Poses and PoseBusters benchmarks, delivering 14.5% and 14.2% improvements, respectively, over the next best model. In the pocket-conditional cofolding regime, Pearl delivers $3.6\times$ improvement on a proprietary set of challenging, real-world drug targets at the more rigorous RMSD < 1 \r{A} threshold. Finally, we demonstrate that model performance correlates directly with synthetic dataset size used in training.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper introduces Pearl, an AI model that predicts how a small molecule (a potential medicine) fits into a protein in 3D. Think of the protein as a lock and the small molecule as a key. Pearl tries to place every atom of the key into the lock in just the right way. This matters because finding good fits can help scientists design new drugs faster and more accurately.
What questions were the researchers trying to answer?
In simple terms, they asked:
- How can we predict the 3D “pose” (position and shape) of a drug molecule when it binds to a protein, even when we don’t have tons of lab data?
- How can we make sure the predicted poses are not only close to the real ones but also physically possible (no impossible overlaps or broken bonds)?
- Can we give the model hints (like a known pocket shape) and get even better results?
- Does training on lots of realistic, computer-generated examples help the model learn better?
How did they do it?
The team built Pearl with three big ideas in mind. Here’s what they did, in everyday language:
- More (and smarter) training data:
- Real 3D protein–ligand structures from experiments are rare and biased toward popular targets.
- So they created lots of “synthetic” examples using physics-based simulations—like making many realistic practice puzzles—so Pearl could learn from a wider variety of situations.
- A model that respects 3D space:
- Pearl includes a special “equivariant” module. Translation: if you rotate the molecule in 3D, the model’s understanding rotates with it. Imagine turning a real lock-and-key in your hand—the fit should look the same from any angle. This helps Pearl learn efficiently and generalize better.
- They use a diffusion process for generation, which is like un-blurring an image step by step until a clear 3D structure appears.
- Helpful hints during use (controllability):
- Pearl can work in two modes:
- Unconditional: it only gets the protein’s sequence and the drug’s shape/connection info, and figures out the pose from scratch.
- Conditional (pocket-aware): it also gets guidance, like a known binding pocket or a related structure, and uses that as a template. This is like giving it a blueprint to follow.
- Pearl can also be “steered” to enforce certain rules or to increase the diversity of poses it generates.
- Training strategy and engineering:
- They used a step-by-step training curriculum, starting simple and getting more complex.
- They kept calculations efficient and stable with careful use of mixed-precision math and fast GPU kernels—basically, they made it run faster without losing accuracy.
Key terms explained:
- Protein–ligand “pose”: the exact 3D position and shape of the small molecule inside the protein pocket.
- RMSD (Root Mean Squared Deviation): a score for how close the predicted pose is to the ground truth. Lower is better. You can think of it as the average distance between where atoms should be and where the model put them.
- Physically valid: the pose doesn’t break basic chemistry rules (no impossible overlaps, bond lengths make sense, etc.).
What did they find?
Pearl performed better than other leading models (including AlphaFold 3 and several open-source systems) on key tests. Here’s the big picture:
- Strong accuracy on public benchmarks:
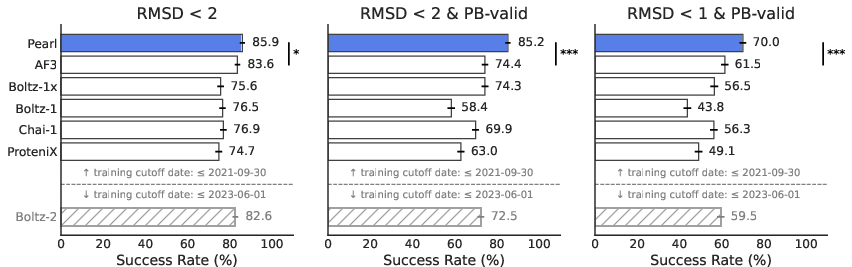
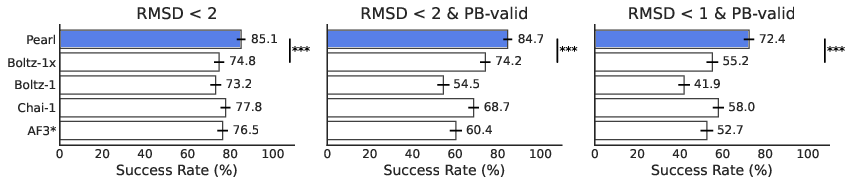
- On Runs N’ Poses and PoseBusters (well-known test sets), Pearl correctly predicted about 85% of poses at the standard accuracy level.
- It also kept those poses physically sensible—its success barely dropped after strict physics checks, which means it mostly makes realistic predictions, not just “looks-right” guesses.
- Better at high-precision poses:
- When the tests used tougher standards (stricter RMSD and physics checks), Pearl still stayed strong while other models dropped more. This matters because drug designers often need very precise poses to improve potency.
- Generalizes to new, real-world targets:
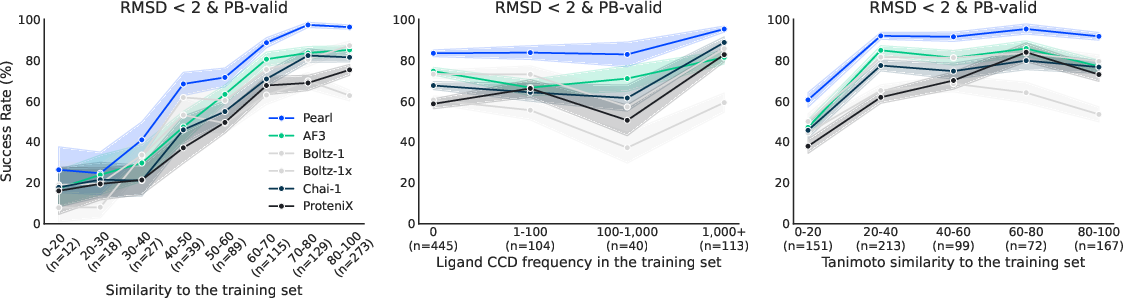
- On a separate, more challenging internal set (with less similarity to what it saw during training), Pearl still clearly outperformed other open models. This suggests it learned general rules rather than memorizing examples.
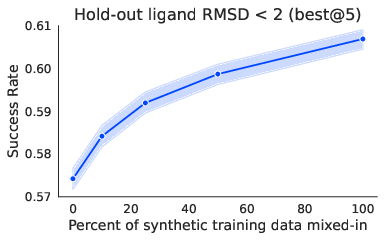
- Scales with more synthetic data:
- The more high-quality synthetic examples they mixed into training, the better Pearl performed. This shows synthetic data can effectively fill gaps when lab data is limited.
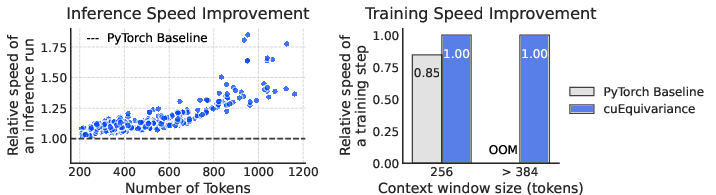
- Efficient and practical:
- With smart engineering, Pearl runs faster and uses memory better, which makes it more practical to use in real projects.
Why does it matter?
- Faster, smarter drug discovery:
- Knowing how a potential drug fits into a protein helps scientists decide which molecules to make and test. That can save time and money and speed up finding new treatments.
- Reliable guidance for chemists:
- Pearl’s poses are both accurate and physically realistic, which reduces the risk of being misled by “pretty but impossible” structures.
- Better use of limited data:
- Biology doesn’t have as much labeled data as text or images. Pearl shows that combining physics-aware AI with lots of realistic synthetic examples can still achieve state-of-the-art results.
- A blueprint for scientific AI:
- The techniques here—equivariant models that respect real-world geometry, carefully controlled inference, and synthetic data at scale—could help in other areas of science where data is scarce but physics matters.
In short, Pearl is a strong step toward AI tools that can place every atom in the right spot, giving scientists clearer, more trustworthy 3D insights to design better medicines.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a single, concrete list of gaps and open questions the paper leaves unresolved. Each item is phrased to enable targeted follow-up by future researchers.
- Synthetic data generation: The paper does not fully describe how synthetic protein–ligand complexes were generated (e.g., physics engine, scoring/validation criteria, protonation/tautomer rules, conformer sampling, pocket preparation), making it hard to assess fidelity, bias, and reproducibility.
- Synthetic data quality control: No quantitative analysis of the chemical/structural realism of synthetic poses (e.g., torsion strain distributions, sterics, metal coordination, hydrogen-bond geometries, water-mediated interactions, protonation states) or of how these differ from experimental structures.
- Synthetic data mixing strategy: The optimal mixing ratio between experimental and synthetic data, and whether diminishing returns or adverse effects (e.g., hallucination, bias amplification) emerge at scale, is not studied for the flagship model.
- Synthetic data domain bias: The composition of synthetic ligands (chemotypes, ring systems, macrocycles, charge states, sizes) and pockets (protein families, membrane vs soluble, metalloenzymes) is not reported, leaving open whether the synthetic corpus introduces or amplifies biases.
- Scaling law characterization: While performance correlates with synthetic dataset size for a smaller model, there is no formal scaling-law analysis across model size, data size, and compute; the generality of the trend and the presence of data/model/compute breakpoints remain unknown.
- Architectural ablations: The contribution of the SO(3)-equivariant diffusion module to accuracy and physical validity is not isolated via head-to-head ablations against non-equivariant versions under identical training conditions.
- Trunk vs diffusion contributions: The relative impact of trunk design choices (e.g., triangle multiplication, pairwise representation) versus diffusion module on accuracy, validity, and efficiency is not quantified.
- Mixed precision effects on accuracy: The bf16/fp32 mixed-precision strategy is profiled for speed and memory, but not systematically for numerical stability and accuracy across varied inputs (large complexes, long sequences, metals, charged ligands).
- Inference-time guidance details: The “optional guidance and steering” methods are not specified (e.g., constraint types, implementation, guidance strength schedule), nor is their effect on accuracy/diversity/validity quantified.
- Confidence and ranking: Practical pose selection remains unaddressed—best@k uses oracle selection against ground truth; calibrated confidence scores, uncertainty quantification, and robust ranking (without ground truth) are not evaluated.
- Diversity–accuracy trade-offs: The ability to “modulate pose diversity” is claimed but not measured; diversity metrics, coverage of plausible binding modes, and the trade-off against accuracy/validity are not characterized.
- Robustness of conditional mode: Sensitivity analyses for pocket-aware conditioning are missing—how performance changes under imperfect templates (misaligned, partially missing residues, alternative conformations, wrong apo/holo state) or noisy pocket residue sets.
- Template retrieval and alignment: The template search and alignment strategy (features, thresholds, MSA usage, cross-chain alignment, inclusion of non-polymeric components) is not described, and its contribution to performance is not quantified.
- Template-induced bias: Whether conditioning on related ligands risks information leakage or narrows exploration (e.g., mode collapse to known pockets) is not studied; guardrails for template similarity thresholds are undefined.
- Generalization beyond protein–ligand: Pearl is positioned as a general biomolecular cofolding model, but benchmarks and analyses are restricted to protein–ligand; performance on nucleic acids, peptides, glycans, lipids, cofactors, multi-ligand complexes, and multimeric assemblies is not evaluated.
- Coverage of challenging chemistries: Systematic evaluation on macrocycles, covalent inhibitors, highly flexible ligands, zwitterions, polycationic/polyanionic compounds, and metal-chelating ligands is missing.
- Protonation/tautomers/stereochemistry: The handling of ligand protonation states, tautomerism, and stereochemical assignments at inference and training time is unclear and not stress-tested for impact on pose accuracy and validity.
- Waters and ions: Treatment of crystallographic waters and ions (especially catalytically essential ones) during training/inference and their contribution to pose fidelity are not addressed.
- Protein flexibility scope: While induced-fit is discussed, the extent to which Pearl models larger-scale conformational changes (e.g., domain movements, loop disorder/order transitions) is not quantified; no analyses by induced-fit magnitude.
- Membrane proteins and difficult targets: Performance on membrane-bound targets, GPCRs, transporters, intrinsically disordered regions, and low-resolution/recalcitrant systems is not stratified.
- Physical validity beyond PoseBusters: PoseBusters checks are helpful but limited; deeper physics metrics (e.g., energy-based strain, rotamer plausibility, clash energies, interaction geometry distributions, MD relaxation stability) are not applied.
- Affinity and SAR utility: The link between high-resolution pose accuracy and downstream utility (e.g., enrichment in virtual screening, correlation with binding affinities across congeneric series, potency prediction improvements) is not demonstrated.
- Prospective validation: No prospective, blinded wet-lab validation is presented (e.g., predicting poses for yet-to-be-solved complexes and later verifying experimentally).
- Pose selection vs medicinal chemistry thresholds: Although stricter RMSD thresholds are reported, practical thresholds and error modes that affect medicinal chemistry decisions (e.g., key interaction recovery rates, ring flip detection, subpocket misassignment) are not systematically quantified.
- Best@k comparability: Different baselines use varying selection regimes (e.g., max-confidence), complicating comparability; a unified evaluation with standardized sampling counts and selection protocols is missing.
- Data leakage and deduplication: Despite date cutoffs, rigorous leakage controls (e.g., homology-based deduplication, ligand/pocket near-neighbor removal) are not fully described; the extent of memorization vs generalization is inferred but not proven via controlled experiments.
- InternalXtals reproducibility: Proprietary dataset results cannot be externally replicated; a public analog or release of benchmark criteria and selection methodology would enable independent verification.
- Compute accessibility: Training/evaluation relies on H100/H200 GPUs and optimized kernels; guidance for scaling to resource-limited environments (smaller models, pruning/distillation, low-memory inference) is not provided.
- Throughput and cost: Inference-time costs, throughput per complex, and scalability for large libraries (e.g., thousands of ligands per target) are not reported.
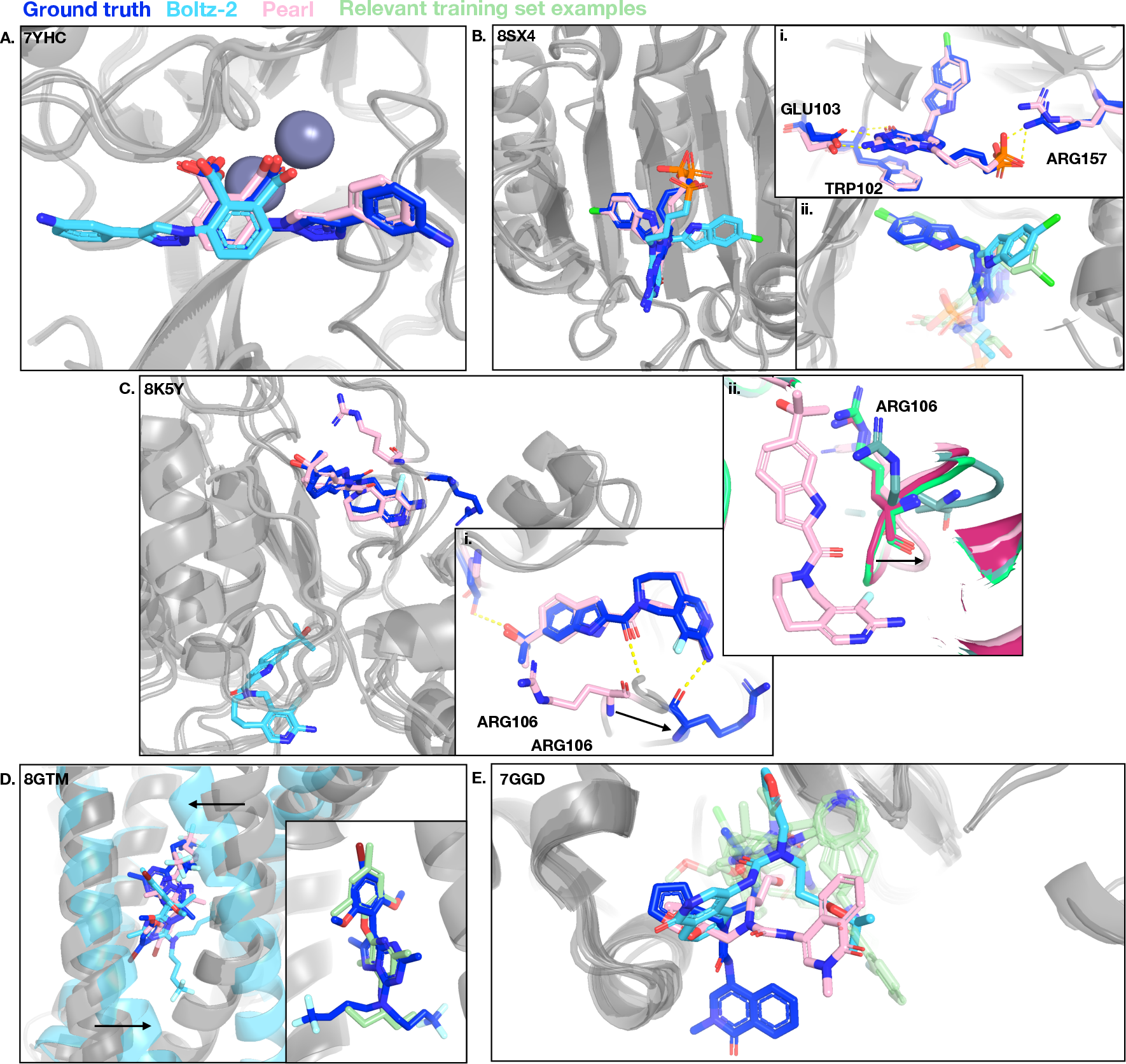
- Failure mode taxonomy: Case studies suggest memorization and subpocket biases, but there is no systematic taxonomy or quantitative failure analysis across datasets to inform targeted model improvements.
- Induced bias from template frequency: Models may prefer frequently occupied pockets; experiments perturbing template distributions to measure bias strength and mitigation strategies are absent.
- Training curriculum ablations: The five-stage curriculum’s individual contributions are not isolated; removing or reordering stages to test necessity/sufficiency is an open experiment.
- Noise schedule and diffusion details: Core diffusion settings (noise schedule, denoising steps, parameterization) and their impact on accuracy/validity/speed are not documented or ablated.
- Model size and parameters: The parameter count, memory footprint, and context limits are not reported, hindering reproducibility and scaling studies.
- Multi-ligand and stoichiometry: Capability to model complexes with multiple ligands/cofactors simultaneously, and effects on accuracy and validity, are untested.
- Cross-target selectivity: The model’s ability to distinguish on-target pockets from off-target sites and predict selective binding modes is not evaluated.
- Alignment between predicted pocket conformations and experimental variability: How often predicted conformations fall within the distribution of experimentally observed pocket states (e.g., ensemble coverage) is not measured.
- Integration with physics-based refinement: The benefit of post-prediction energy minimization or MD refinement (and whether it improves or degrades Pearl’s poses) is not assessed.
- Error calibration and reliability diagrams: Confidence calibration (e.g., reliability diagrams for RMSD prediction) and uncertainty estimates are not provided, limiting safe deployment.
- Robustness to input noise: Sensitivity to errors in protein sequence, ligand topology, chain breaks, missing residues, and ambiguous atom typing is not studied.
- Template-free discovery: For truly novel pockets without templates/MSAs, the unconditional mode’s limits and failure modes are not comprehensively profiled across pocket novelty spectra.
- Fair licensing comparisons: Some baselines (e.g., AF#13) were excluded in certain settings due to licensing, leaving a gap for fully fair, apples-to-apples comparisons under permissive configurations.
Practical Applications
Immediate Applications
The following applications can be deployed now using the methods and findings reported (e.g., SO(3)-equivariant cofolding, multi-chain templating with non-polymeric components, controllable inference, and synthetic-data–augmented training). Each item lists sectors, potential tools/workflows, and key dependencies.
- Augment or replace rigid docking in virtual screening with cofolding poses
- Sectors: healthcare, pharma/biotech, software (computational chemistry)
- What: Use Pearl’s unconditional cofolding to generate 20 diverse poses per ligand and select best@k for triage; re-rank or filter classical docking outputs to reduce false positives/negatives and induced-fit failures.
- Tools/workflows: Pipeline nodes for KNIME/Nextflow; RDKit + Pearl for pose generation; PoseBusters-like validity checks to filter strained poses.
- Assumptions/dependencies: Access to the model/API and GPUs; reliable ligand topology (tautomer/protonation states); data governance for proprietary structures.
- Pose-seeded physics workflows (cut cost of MD/IFD/FEP)
- Sectors: healthcare, pharma/biotech
- What: Initialize induced-fit docking, short MD relaxations, and alchemical FEP with physically plausible Pearl poses to reduce sampling time and increase convergence rates.
- Tools/workflows: Schrodinger/MOE/AMBER/GROMACS pipelines; PLIF/IPFP generation for interaction benchmarking.
- Assumptions/dependencies: Accurate force fields/parameters (e.g., metals/cofactors); standardized protonation and ionization states.
- Pocket-conditional cofolding for SAR cycles where a structure exists
- Sectors: healthcare, pharma/biotech
- What: Use multi-chain templating (including cofactors and reference ligands) to guide design sprints for analog series, scaffold hops, or series migration across isoforms.
- Tools/workflows: Template assembly from apo/holo PDB/Cryo-EM; conditional generation plus guidance to enforce interaction motifs; best@k selection with medicinal chemistry review.
- Assumptions/dependencies: Availability/accuracy of reference pockets; pocket residue definition; sensitivity to template quality.
- 3D feature generation to improve QSAR/ML potency models
- Sectors: healthcare, pharma/biotech, software
- What: Generate consistent 3D poses per ligand–target pair to derive 3D descriptors, interaction fingerprints, and contact maps feeding hybrid QSAR models.
- Tools/workflows: RDKit/DeepChem feature extraction; PLIP/ODDT for interaction fingerprints; model stacking with classical descriptors.
- Assumptions/dependencies: Target coverage; avoidance of propagating wrong poses—use PB-valid constraints and confidence gating.
- Multi-target selectivity and off-target risk assessment
- Sectors: healthcare, pharma/biotech, safety/toxicology
- What: Run conditional cofolding across homologs and off-target panels (e.g., hERG kinases) to rapidly compare binding modes and flag liabilities.
- Tools/workflows: Batch cofolding with homolog templates; cross-target PLIF overlap and differential contact scoring.
- Assumptions/dependencies: Quality of homolog templates; pose comparability across targets; domain expertise to interpret subtle interaction shifts.
- Rapid hypothesis support for structural biology and assay triage
- Sectors: healthcare, academia
- What: Generate binding-mode hypotheses to guide construct design, mutagenesis, and crystallization; reconcile ambiguous electron density; reject strained or implausible poses early.
- Tools/workflows: Density-fitting validation aided by PB-valid screens; mutational hotspot prediction from contact maps.
- Assumptions/dependencies: Experimental follow-up; proper ligand protonation/tautomer enumeration.
- Integration into closed-loop DMTA with generative chemistry
- Sectors: healthcare, pharma/biotech, software/AI platforms
- What: Score generative candidates by cofolding poses plus interaction objectives (e.g., hydrogen-bond geometry, metal coordination), steer generation via pose-based constraints.
- Tools/workflows: Pearl + generative models (graph/SMILES diffusion) with pose-constrained scoring; Bayesian optimization/active learning on batch assay data.
- Assumptions/dependencies: Careful objective design to avoid gaming; automated sanitization of unrealistic chemistries.
- Metalloenzyme and cofactor-aware modeling out-of-the-box
- Sectors: healthcare, industrial biocatalysis
- What: Use non-polymeric templating to include metals (Zn2+, Mg2+, Fe) and cofactors (NAD+, FAD) to improve placement and interaction realism.
- Tools/workflows: Template builders that include cofactors; chemistry checks for coordination geometries.
- Assumptions/dependencies: Correct cofactor states; param set alignment for downstream physics.
- Benchmarking and internal model evaluation with stronger validity criteria
- Sectors: academia, industry R&D, standards organizations
- What: Adopt RnP/PoseBusters-like best@k with PB-valid filters to audit in-house pipelines and report reproducible, physically valid progress.
- Tools/workflows: CI dashboards; standardized seed/config logging; ablation reporting (synthetic data proportions).
- Assumptions/dependencies: Access to curated benchmark splits; consistent train/test cutoffs.
- Educational and communication tools for medicinal chemistry and biochemistry
- Sectors: education, daily life (professional training)
- What: Interactive 3D notebooks/apps showing predicted complexes, highlighting key contacts and “what-if” side-chain/ligand modifications.
- Tools/workflows: WebGL/py3Dmol viewers; embedded pose-generation widgets in Jupyter.
- Assumptions/dependencies: Simplified UIs; compute quotas for classrooms.
- Secure, on-prem or cloud deployment patterns
- Sectors: software, regulated pharma IT
- What: Containerized inference servers with H100-class GPUs; SDKs for batch screening; audit trails for GxP-adjacent environments.
- Tools/workflows: Kubernetes/HPC integration; workflow managers; role-based access controls.
- Assumptions/dependencies: Licensing and model availability; GPU capacity and cost.
Long-Term Applications
These applications will benefit from additional research, scaling, validation, or ecosystem development (e.g., expanded training sets, prospective studies, regulatory acceptance).
- End-to-end generative design with co-optimized pose fidelity and properties
- Sectors: healthcare, pharma/biotech, software
- What: Jointly generate molecules and poses under target- and pocket-conditional constraints with physics-informed guidance for potency, selectivity, and developability.
- Tools/workflows: Multi-objective RL/diffusion with pose guidance; in-loop Pearl scoring; downstream ADMET predictors.
- Assumptions/dependencies: Robustness against reward hacking; larger and more diverse training data (including negative/bad-binder examples).
- Variant-aware and patient-specific modeling for precision medicine
- Sectors: healthcare
- What: Predict differential binding modes/affinities across patient mutations (oncology, antimicrobial resistance) to guide therapy selection or resistance-avoidant designs.
- Tools/workflows: Cohort variant sweeps; ΔΔG pipelines seeded by cofolding poses; clinical decision support integration.
- Assumptions/dependencies: High-quality variant annotations; prospective validation; regulatory frameworks for clinical use.
- Enzyme–substrate and biocatalyst design for green chemistry and energy
- Sectors: energy, industrial chemistry, sustainability
- What: Model enzyme–substrate complexes and transition-state analogs to guide catalytic pocket engineering for biofuels, fine chemicals, and CO2 utilization.
- Tools/workflows: Conditional cofolding of enzyme–substrate; coupling to QM/MM or EVB for barrier estimates; directed evolution targeting predicted hotspots.
- Assumptions/dependencies: Extension of training coverage to enzymatic chemistries and transition-state mimics; careful treatment of reactive states.
- Regulatory-grade in silico evidence pipelines
- Sectors: healthcare, policy/regulation
- What: Standardized, auditable computational evidence packages where physically valid cofolding poses seed physics, structure-based SAR, and mechanism-of-action narratives in submissions.
- Tools/workflows: SOPs for pose generation/selection; uncertainty quantification; model cards and data provenance for synthetic datasets.
- Assumptions/dependencies: Consensus standards for pose validity and reporting; prospective benchmarks accepted by regulators.
- Automated, robotic Design–Make–Test–Analyze with active learning
- Sectors: healthcare, robotics, software
- What: Integrate cofolding-based ranking into autonomous labs that perform synthesis/assays, using active learning to refine models and pocket hypotheses.
- Tools/workflows: Closed-loop orchestration (ChemOS/ACLS-like); on-the-fly templating updates; experiment design to disambiguate binding modes.
- Assumptions/dependencies: Reliable synthesis and assay automation; robust uncertainty estimation; budget and throughput alignment.
- Polypharmacology and network-level design
- Sectors: healthcare, systems pharmacology
- What: Simultaneously optimize binding modes across target panels to engineer desired polypharmacology profiles while avoiding off-target liabilities.
- Tools/workflows: Multi-target conditional cofolding; cross-target PLIF harmonization; Pareto front exploration.
- Assumptions/dependencies: Comprehensive structure coverage; scalable multi-target scoring; data on off-target safety thresholds.
- Expanded modality coverage (RNA, DNA, peptides, macrocycles, PROTACs, covalent binders)
- Sectors: healthcare, biotech
- What: Extend cofolding to additional non-polymeric components and linkage chemistries (e.g., warheads, linkers) with accurate placement and reactivity-aware constraints.
- Tools/workflows: Enriched training sets and synthetic data for new modalities; covalent pose validation; linker conformational sampling guided by cofolding.
- Assumptions/dependencies: Data scarcity for new modalities; specialized validity checks (e.g., covalent geometry rules).
- Distillation and hardware-efficient variants for wider access
- Sectors: software, policy (access equity)
- What: Smaller, distilled models or on-demand cloud services enabling SMEs and academic labs to run cofolding at scale without H100-class hardware.
- Tools/workflows: Knowledge distillation; quantization; serverless inference endpoints.
- Assumptions/dependencies: Maintaining accuracy/validity under compression; sustainable cost models.
- Data and policy ecosystem for synthetic training data
- Sectors: policy, academia, industry consortia
- What: Standards for provenance, bias auditing, and disclosure of synthetic structures; shared repositories enabling reproducible scaling studies.
- Tools/workflows: Model/data cards; benchmark registries with locked splits; governance frameworks for synthetic data generation pipelines.
- Assumptions/dependencies: Cross-stakeholder agreements; privacy/IP safeguards.
- Environmental health and safety use-cases
- Sectors: public health, toxicology
- What: Predict binding modes for safety-relevant targets (e.g., nuclear receptors, ion channels) for early tox de-risking of consumer chemicals and drugs.
- Tools/workflows: Panel cofolding + tox informatics; integration with in vitro new approach methodologies (NAMs).
- Assumptions/dependencies: Coverage of safety targets; validation against reference tox datasets.
- Real-world impact on daily life via faster, cheaper therapies
- Sectors: daily life (societal benefits)
- What: Shorter discovery cycles and higher success probabilities leading to earlier availability of treatments in infectious disease, oncology, CNS, and rare diseases.
- Tools/workflows: Portfolio analytics linking cofolding-enabled acceleration to time-to-clinic metrics; health-economic models.
- Assumptions/dependencies: Translation of in silico gains to clinical outcomes; multi-factor pipeline efficiencies beyond binding pose prediction.
Cross-cutting assumptions and dependencies (common to many items)
- Model availability and licensing terms; ability to deploy on-prem or via API.
- Compute access and cost (H100/H200-class GPUs); potential need for distilled variants.
- Data quality: ligand standardization, correct protonation/tautomer states, accurate templates/MSAs, and careful curation of synthetic data.
- Human-in-the-loop expertise for pose interpretation and selection; organizational adoption of best@k and PB-valid practices.
- Prospective validation and benchmarking consistency to ensure generalization beyond PDB-like cases.
Glossary
- AlphaFold #13: A state-of-the-art deep learning model for biomolecular cofolding that generalizes protein folding to many molecule types. "AlphaFold #13 (AF3) \cite{abramson2024accurate} generalized protein folding models to nearly all molecule types in the Protein Data Bank (PDB)~\cite{wwpdb_stats_2025}, inspiring a new generation of cofolding models, such as"
- apo: The unbound conformational state of a protein without a ligand. "a ligand-bound conformational state, in contrast to the unbound (apo) state."
- bfloat16 (bf16): A 16-bit floating-point format with an 8-bit exponent used for efficient mixed-precision computation. "Pearl employs a conservative bfloat16 (bf16) mixed-precision strategy."
- binding pocket: The 3D region of a protein where a ligand binds, often undergoing conformational changes. "Crucially, these cofolding models adapt the protein binding pocket geometry to different ligands, accounting for flexibility more fundamentally than previous approaches."
- CASP14: A community benchmark for protein structure prediction accuracy. "most notably when AlphaFold #12 achieved near-experimental accuracy on the CASP14 challenge on protein monomers"
- cofactor: A non-protein chemical component required for protein function (e.g., metal ions, small molecules). "proteins, ligands, nucleic acids, and other cofactors, predicting structures directly from their sequences and chemical topologies."
- cofolding: Joint prediction of the 3D structure of interacting biomolecules (e.g., protein–ligand complexes). "a foundation model for proteinâligand cofolding at scale."
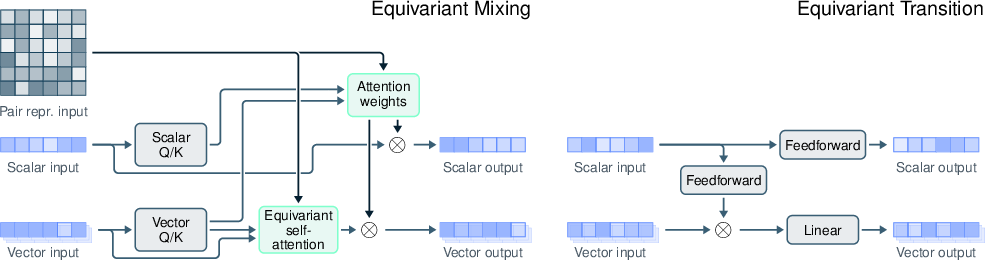
- curriculum training: A training strategy that progressively increases task complexity to improve generalization. "including curriculum training and an architecture that encompasses an SO(3)-equivariant diffusion module."
- cuEquivariance: An NVIDIA library providing optimized GPU kernels for equivariant operations. "We use optimized kernels from cuEquivariance (v0.6.0) \cite{nvidia_cuequivariance_github} and a custom CUDA LayerNorm kernel~\cite{bytedance2025protenix} to accelerate key trunk operations."
- denoising diffusion probabilistic models: Generative models that learn to reverse a noise-adding process to sample structured outputs. "which are often denoising diffusion probabilistic models~\cite{diffusion}."
- diffusion module: A model component that predicts 3D coordinates via a diffusion-based generative process. "architectures that incorporate an SO(3)-equivariant diffusion module to inherently respect 3D rotational symmetries"
- equivariant transformer (EqT): A transformer architecture that enforces geometric equivariance (e.g., under rotations). "constructed from equivariant transformer (EqT) blocks (Figure~\ref{fig:eqt})."
- fp32: Standard 32-bit floating-point precision used for numerical stability. "all model weights remain in full fp32 precision to ensure stability."
- holo-like: A ligand-bound conformational state of a protein, used to provide realistic pocket context. "a coherent, “holo-like” pocket environment---that is, a ligand-bound conformational state, in contrast to the unbound (apo) state."
- induced fit: Protein conformational changes that occur upon ligand binding. "induced fit---the protein conformational changes that may occur upon binding~\cite{inducedfit1,inducedfit2,miller2021inducedfitdocking}"
- InternalXtals: A proprietary benchmark of internal crystal structures for real-world generalization testing. "InternalXtals dataset consists of 111 structures from a variety of internal programs."
- LayerNorm: A normalization operation applied within neural network layers to stabilize training. "LayerNorm \cite{ba2016layer}"
- ligand docking: Physics-based computational placement of ligands into protein binding sites. "Traditionally, 3D structure prediction has been performed using physics-based ligand docking methods~\cite{rarey_fast_1996,surflex,mcgann_gaussian_2003,gold,friesner_glide_2004,corbeil_variability_2012,venkatraman_flexible_2012,AutoDockVina}."
- ligand strain: Energetic penalty from distorted ligand geometry in a predicted pose. "such as low ligand strain, shape complementarity, and favorable non-covalent interactions."
- monomer distillation data: Single-chain protein data distilled from models/databases to augment training. "monomer distillation data (OpenFold \cite{ahdritz2024openfold} and the AlphaFold Database \cite{varadi2024alphafold})"
- multi-chain templating: Conditioning using templates across multiple chains (and components) to guide generation. "a generalized multi-chain templating system supporting both protein and non-polymeric components"
- multiple sequence alignments (MSAs): Aligned sets of homologous sequences capturing evolutionary signals. "evolutionary information (i.e. MSAs)"
- non-covalent interactions: Physical interactions not involving covalent bonds (e.g., hydrogen bonds, van der Waals). "and favorable non-covalent interactions."
- non-polymeric components: Complex components that are not polymers (e.g., ligands, cofactors). "templates that also include non-polymeric components."
- OpenFold: An open-source protein structure prediction framework used for distillation data. "OpenFold \cite{ahdritz2024openfold}"
- Pairformer: AF3’s trunk module for pairwise representation learning. "a trunk module (e.g., AF3's Pairformer) and a generative structure module, which are often denoising diffusion probabilistic models~\cite{diffusion}."
- PB-valid: PoseBusters physical plausibility validation criterion. "physical plausibility checks from the PoseBusters \cite{buttenschoen2024posebusters} validation suite (PB-valid; Appendix~\ref{app:eval:metrics})."
- PoseBusters: A benchmark and validation suite for assessing pose realism and physical validity. "PoseBusters benchmark."
- Protein Data Bank (PDB): The principal repository of experimentally determined biomolecular structures. "The Protein Data Bank (PDB)~\cite{berman2000protein} offers orders of magnitude less data than domains like text or images"
- root mean squared deviation (RMSD): A metric for average atom-position error between predicted and reference structures. "The primary accuracy metric is ligand root mean squared deviation (RMSD)~\cite{gilson2025assessment}."
- structure-activity relationship (SAR): The relationship between chemical structure and biological activity, used in drug design. "A common approach involves building structure-activity relationship (SAR) models"
- structure-based drug design (SBDD): Designing ligands using target 3D structural information and physical principles. "A key strategy to improve SAR modeling and to accelerate this process is structure-based drug design (SBDD)"
- SuCOS: A combined overlap score used to measure ligand pose similarity. "combined overlap score [SuCOS] of the ligand pose \cite{vskrinjar2025have}"
- Tanimoto similarity: A fingerprint-based measure of chemical similarity between molecules. "Tanimoto similarity < 0.2."
- templating: Providing structural priors via reference templates to guide model predictions. "standard templating approach from protein-only templates"
- triangle multiplication (trimul): A lightweight module for efficient pairwise representation updates in the trunk. "featuring a lightweight triangle multiplication (trimul) module for efficiency~\cite{pairmixer}"
- pocket conditioning: Conditioning generation on known pocket residues or structures to guide sampling. "dual retrieval capabilities of its versatile multi-chain templating system and pocket conditioning enable Pearl to generate structures in two distinct cofolding modes"
- pocket-conditional cofolding mode: A guided generation mode using pocket information or structural priors. "Pearl performance in the (b) unconditional (Runs N' Poses) and (c) pocket-conditional (InternalXtals) cofolding modes"
- unconditional cofolding mode: A generation mode using only sequence/topology without pocket-specific guidance. "In the unconditional cofolding mode, Pearl predicts the complex structure using only the protein's amino acid sequence and the ligand's topology."
Collections
Sign up for free to add this paper to one or more collections.