- The paper presents IntFold as a novel foundation model that integrates a custom FlashAttentionPairBias kernel and modular adapters to achieve high accuracy and controllability in biomolecular structure prediction.
- Benchmarking results on FoldBench show competitive performance on protein monomer, protein-protein, and protein-ligand interactions, with notable improvements in specialized tasks.
- The modular architecture enables efficient target-specific modeling and binding affinity prediction, making IntFold a valuable tool for advancing drug discovery.
IntFold: A Controllable Foundation Model for Biomolecular Structure Prediction
The paper introduces IntFold, a novel foundation model for biomolecular structure prediction, which aims to provide both state-of-the-art accuracy and user-driven controllability. The model leverages a custom attention kernel and modular adapters to achieve high performance on a range of biomolecular interactions and enable specialized applications in drug screening and design. The paper highlights the model's architecture, benchmarking results, applications, data curation strategies, and insights gained during the development process.
Model Architecture and Key Components
The IntFold model (Figure 1) comprises an embedding trunk, a diffusion module, a confidence head, and modular adapters. The embedding trunk processes input sequences and optional MSAs/templates. The diffusion module iteratively generates structure samples, while the confidence head ranks these samples to produce the final prediction. The inclusion of modular adapters allows for specialized downstream tasks like guided folding, target-specific modeling, and affinity prediction.
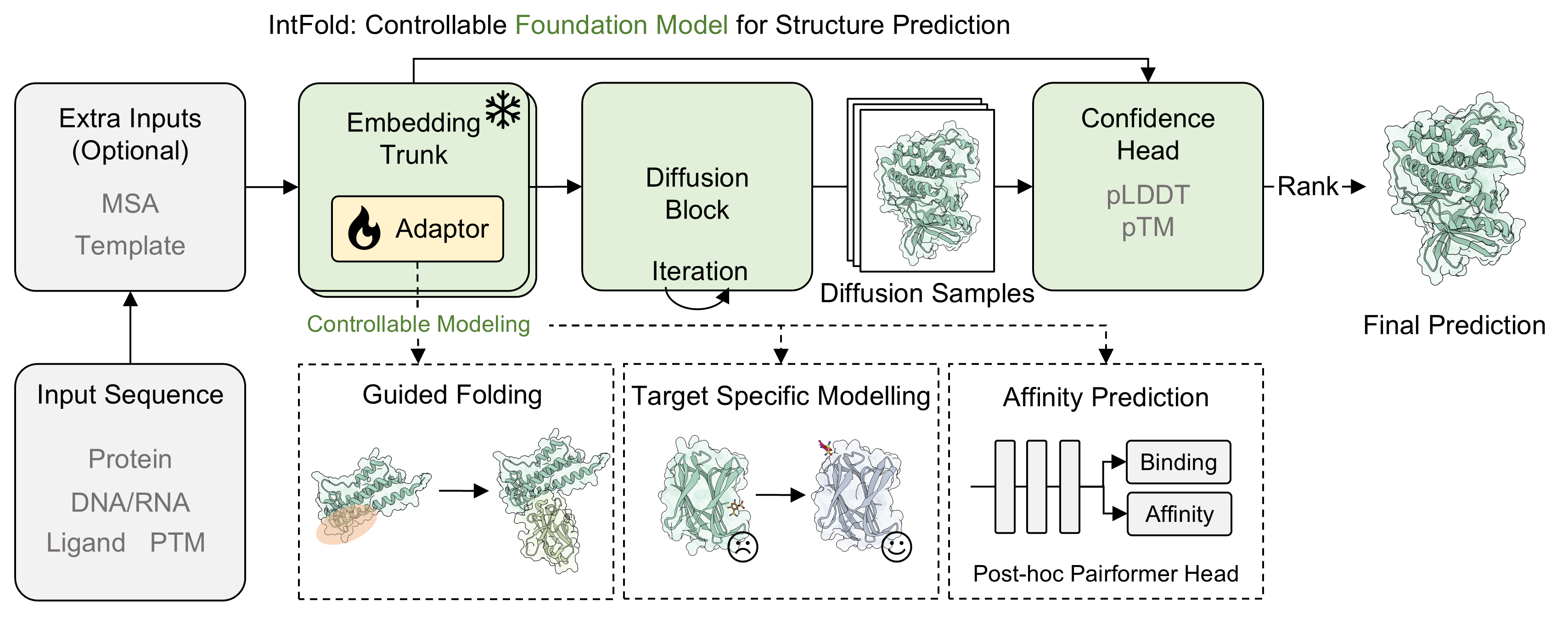
Figure 1: Diagram of the model's architecture illustrating the embedding trunk, diffusion block, confidence head, and modular adapters for specialized tasks.
A key innovation is the custom FlashAttentionPairBias kernel, which outperforms standard industry kernels in both speed and memory usage (Figure 2). This kernel is optimized for handling pair biases in the attention mechanism, leading to improved performance in structure prediction tasks.
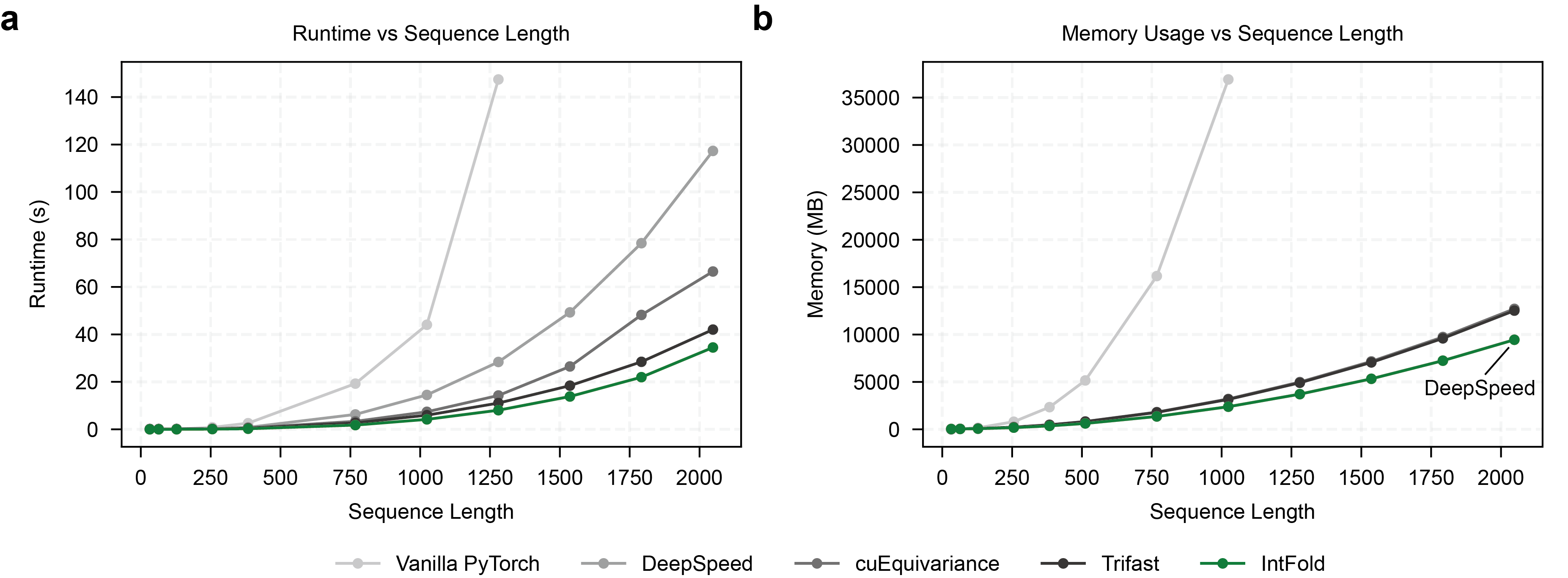
Figure 2: Performance comparison of the custom FlashAttentionPairBias kernel against other implementations, showcasing latency and memory usage advantages.
IntFold's performance was evaluated on the FoldBench benchmark, comparing it against several leading methods, including AlphaFold 3, Boltz-1,2, Chai-1, HelixFold 3, and Protenix. The results demonstrate that IntFold achieves accuracy comparable to AlphaFold 3 on protein monomer prediction, protein-protein interaction prediction, and protein-nucleic acid interaction prediction (Figure 3, Figure 4). Notably, IntFold shows strong performance on antibody-antigen complexes and protein-ligand interactions, modalities critical for therapeutic design (Figure 3, Figure 5).
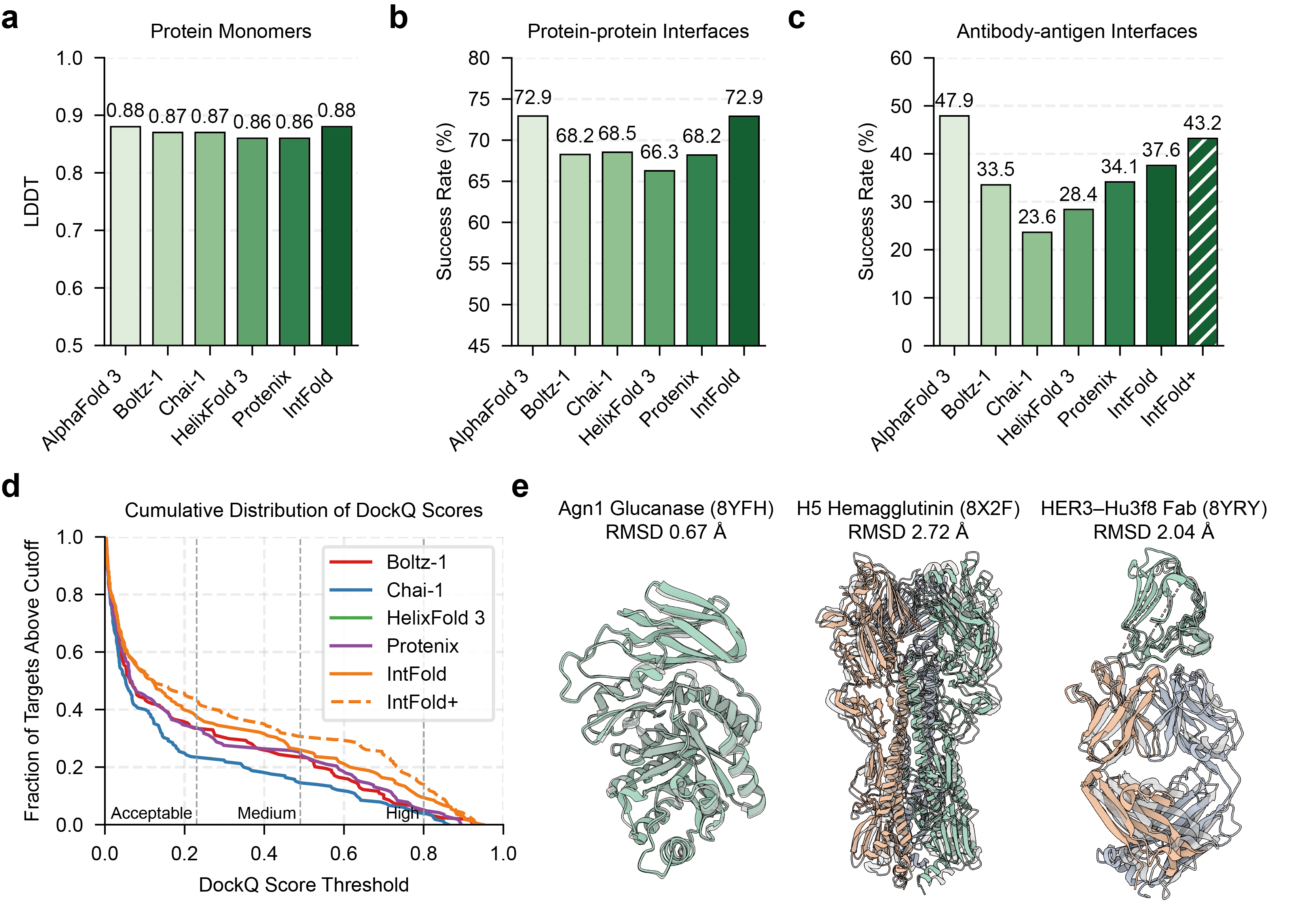
Figure 3: Performance of IntFold on protein monomer and protein-protein complex prediction, showing mean LDDT scores and success rates.
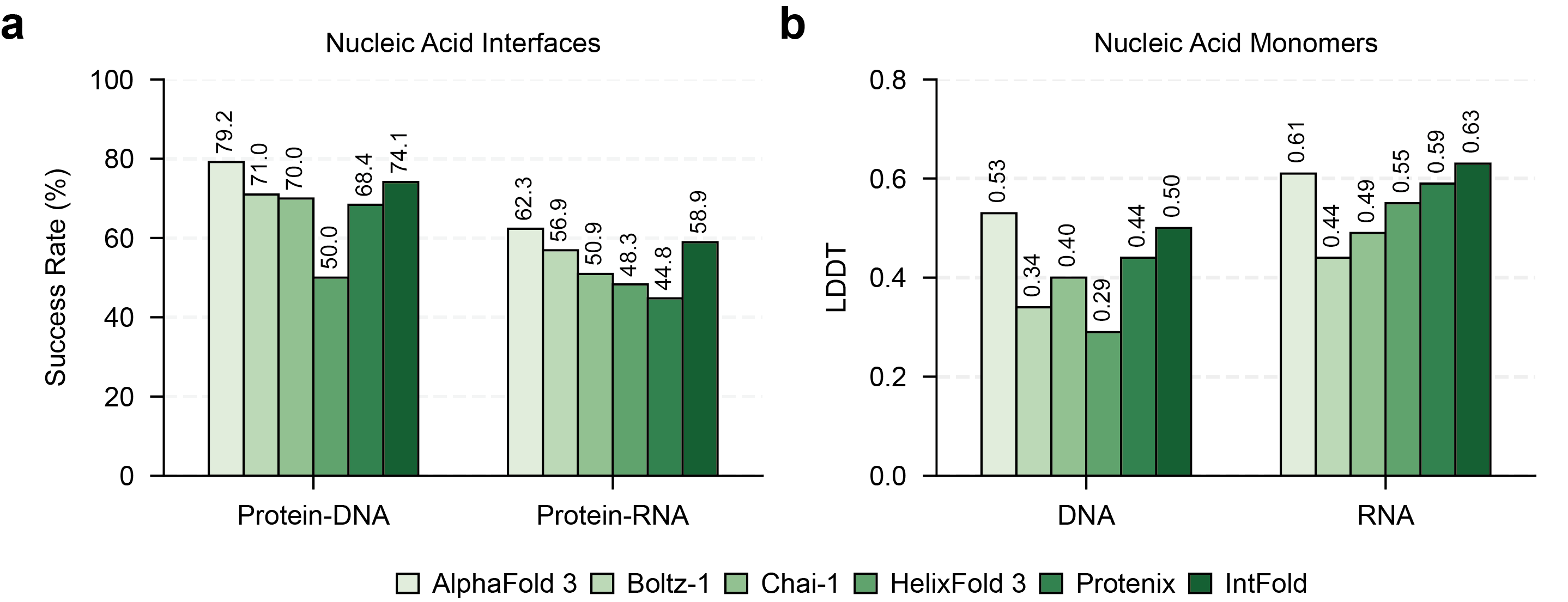
Figure 4: Success rates for protein-nucleic acid interface prediction on FoldBench, including protein-DNA and protein-RNA complexes.
For protein-ligand interactions, IntFold achieves a success rate of 58.5% on FoldBench, placing it as the leading model after AlphaFold 3. Furthermore, IntFold+ improves the success rate to 61.8%, closing the gap to AlphaFold 3 (Figure 5). The model also exhibits strong generalization capabilities, maintaining a significantly higher success rate than other methods for novel ligands.
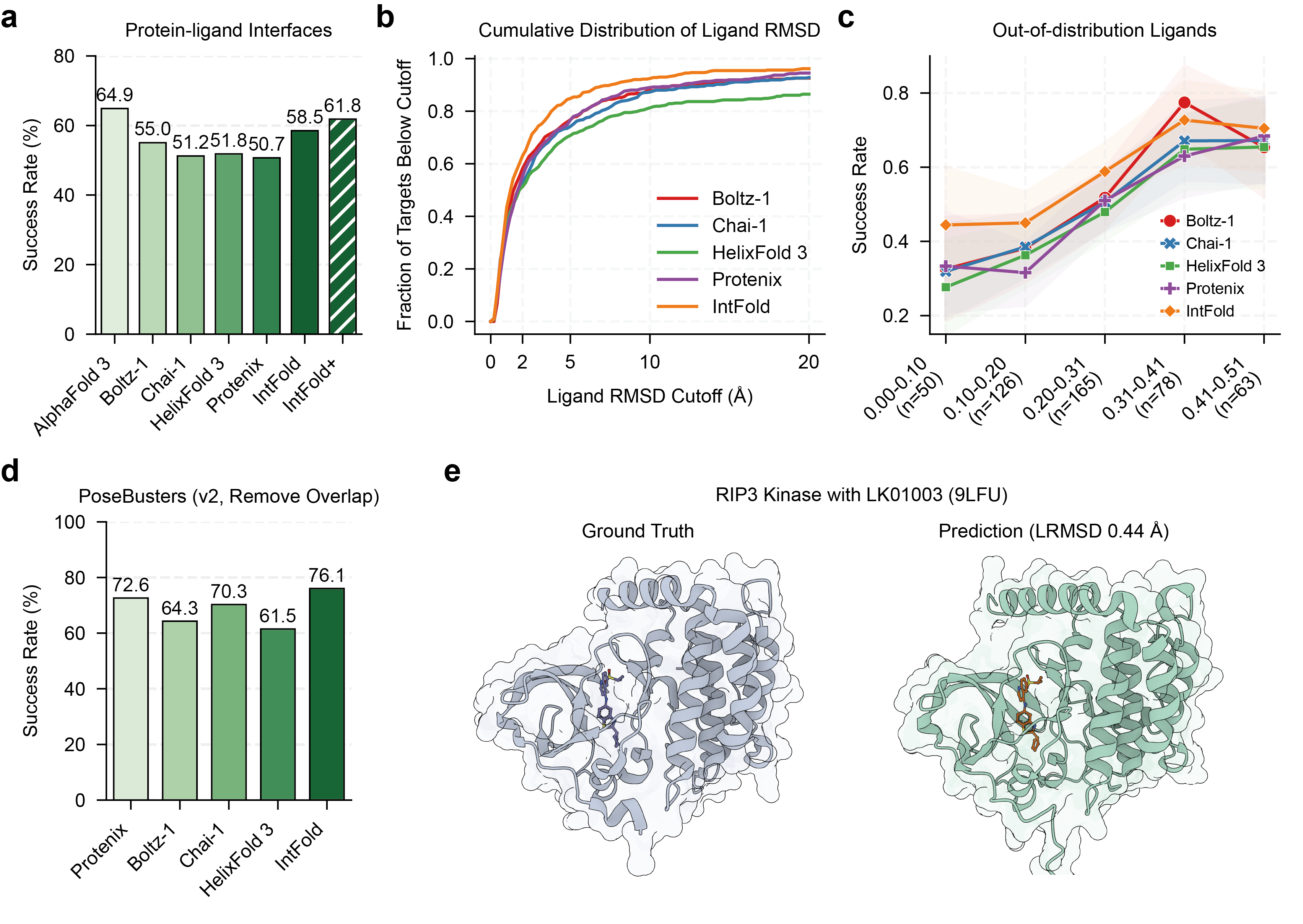
Figure 5: Performance of IntFold on protein-ligand interface prediction, showing success rates, RMSD distributions, and generalization to out-of-distribution molecules.
Specialized Applications Through Modular Adapters
One of IntFold's key strengths lies in its ability to be efficiently specialized for a range of downstream tasks through modular adapters. This is achieved using lightweight, trainable adapters while keeping the large base model frozen. The paper demonstrates the power of this approach with three key applications: improving predictions for specific target families, guiding folding with structural constraints, and predicting binding affinity.
For target-specific modeling, the paper focuses on the kinase family, particularly CDK2. By applying a target-specific adapter, IntFold can capture inhibitor-induced structural shifts that general-purpose models often fail to capture (Figure 6). The fine-tuned model correctly identifies 4 of the 5 allosteric cases in a test set of 40 CDK2 structures, while also maintaining perfect accuracy on the 35 open, inactive-like state structures.
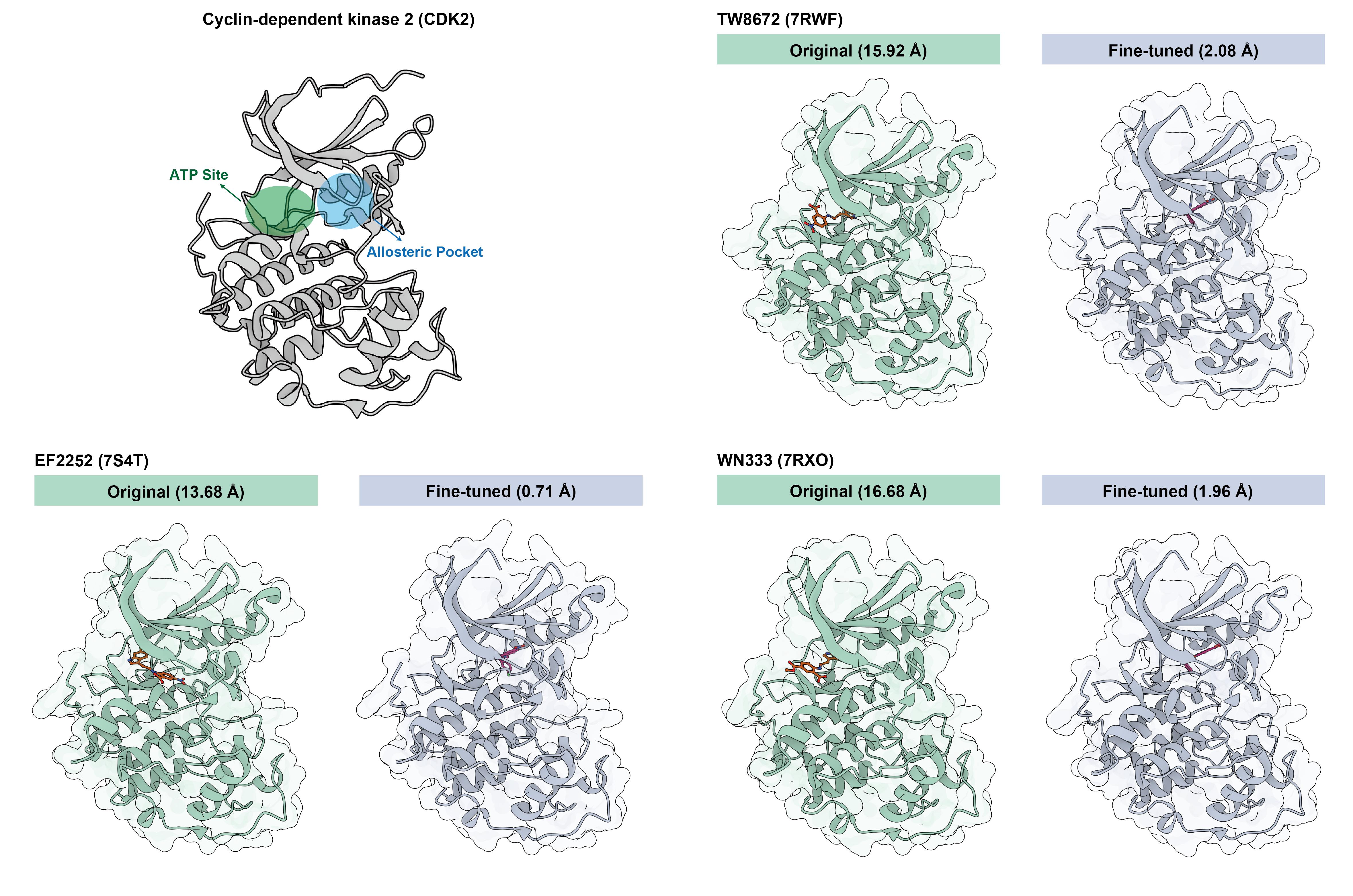
Figure 6: Comparison of IntFold predictions for CDK2 with allosteric inhibitors, showcasing the improvement achieved through fine-tuning with a specialized adapter.
The model also incorporates structural constraints, such as known binding pockets for ligands or specific epitopes for antibodies, leading to improved predictive accuracy. For the PoseBusters dataset, applying constraints boosts the success rate from 79.5% to 89.7% (Figure 7). The effect is even more significant for challenging antibody-antigen interfaces, where the success rate more than doubles from 37.6% to 69.0%.
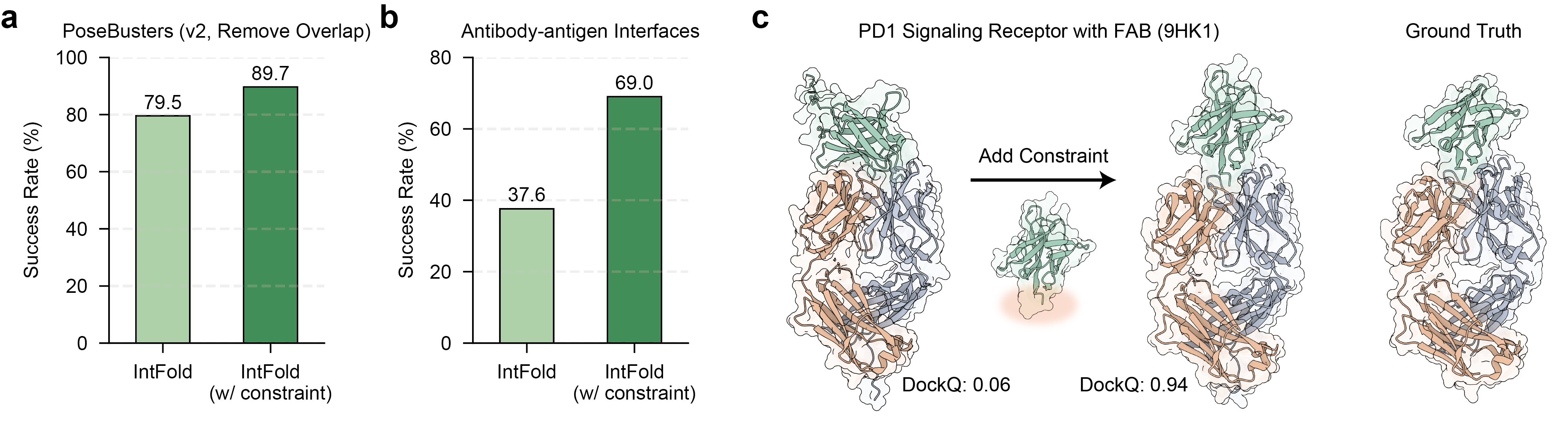
Figure 7: Impact of structural constraints on IntFold's prediction accuracy, demonstrating improved success rates for PoseBusters and antibody-antigen interfaces.
For predicting protein-ligand binding affinity, the model was evaluated on the DAVIS and BindingDB benchmark datasets, with the area under the precision-recall curve (AUPR) as the primary metric. IntFold significantly outperforms a range of existing methods, including both structure-based predictors and various sequence-based approaches (Figure 8).
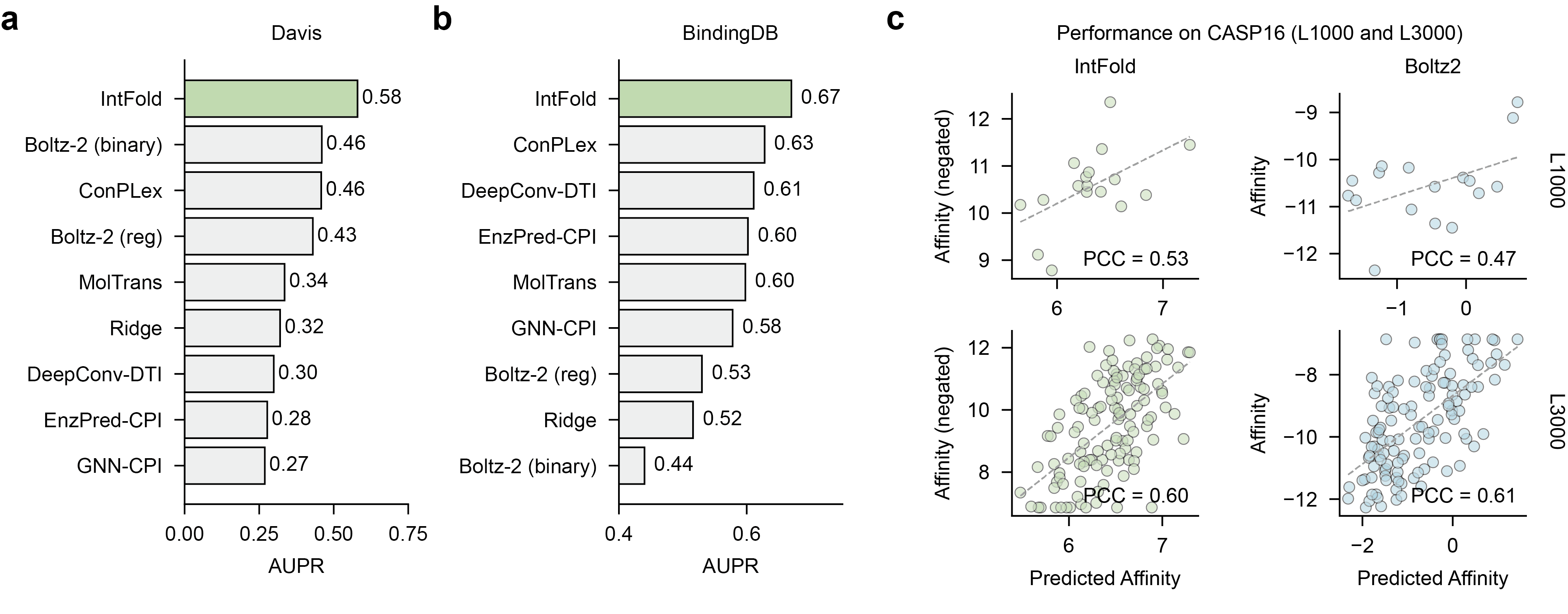
Figure 8: Performance of IntFold on protein-ligand binding affinity prediction, demonstrating superior AUPR scores on the Davis and BindingDB benchmarks.
Data Curation and Training Insights
The paper emphasizes the importance of data scale, diversity, and quality in shaping the performance of foundation models. The data strategy involved integrating high-resolution experimental structures from the PDB with large-scale, high-quality predicted structures through distillation. Specialized datasets were curated for affinity prediction and modeling specific protein families.
During the training process, the authors observed several factors that affect stability, including activation explosion, gradient spikes, parametrization choices, and numerical considerations. To alleviate these issues, they implemented a "skip-and-recover" mechanism and addressed instability related to parametrization and initialization. They found that the diffusion module required full Float32 precision for stable training, while the model's backbone could train effectively with Bfloat16 mixed precision.
Conclusion
IntFold represents a significant advancement in biomolecular structure prediction, offering state-of-the-art accuracy and user-driven controllability. The model's ability to be efficiently specialized for downstream applications through modular adapters makes it a valuable tool for drug discovery and therapeutic design. Future directions include exploring new architectures to mitigate computational complexity, improving predictive accuracy for challenging targets, and expanding functional capabilities to de novo protein design.

