- The paper introduces RSA as a novel method that augments protein language models by retrieving homologous sequences to improve prediction accuracy and reduce computational overhead.
- RSA achieves a 373-fold speedup over traditional MSA methods on secondary structure prediction, outperforming MSA Transformers by at least 5% on average.
- The approach holds promise for accelerating drug discovery and protein engineering by effectively predicting de novo and orphan protein domains without alignment constraints.
Retrieved Sequence Augmentation for Protein Representation Learning
Introduction
Protein representation learning is critical for advancing our understanding of protein functions and structures, which are foundational in many biological processes. The paper "Retrieved Sequence Augmentation for Protein Representation Learning" (2302.12563) presents a novel method called Retrieved Sequence Augmentation (RSA) that enhances protein LLMs by leveraging retrieval-based augmentation without relying on Multiple Sequence Alignments (MSA). RSA provides a faster, efficient alternative that improves predictive performance, particularly in novel protein domains, addressing key challenges associated with computational overhead and the prediction of de novo and orphan proteins.
Methodology
Retrieved Sequence Augmentation (RSA)
RSA is designed to augment protein LLMs by linking query protein sequences with related sequences from a pre-existing database. The RSA framework operates in two main stages: retrieval and augmentation. Firstly, related protein sequences are identified using a dense sequence retriever based on pretrained embeddings, such as those derived from the ESM-1b model. Once retrieved, these sequences are paired with the query protein and fed into the protein LLM to enhance prediction accuracy.
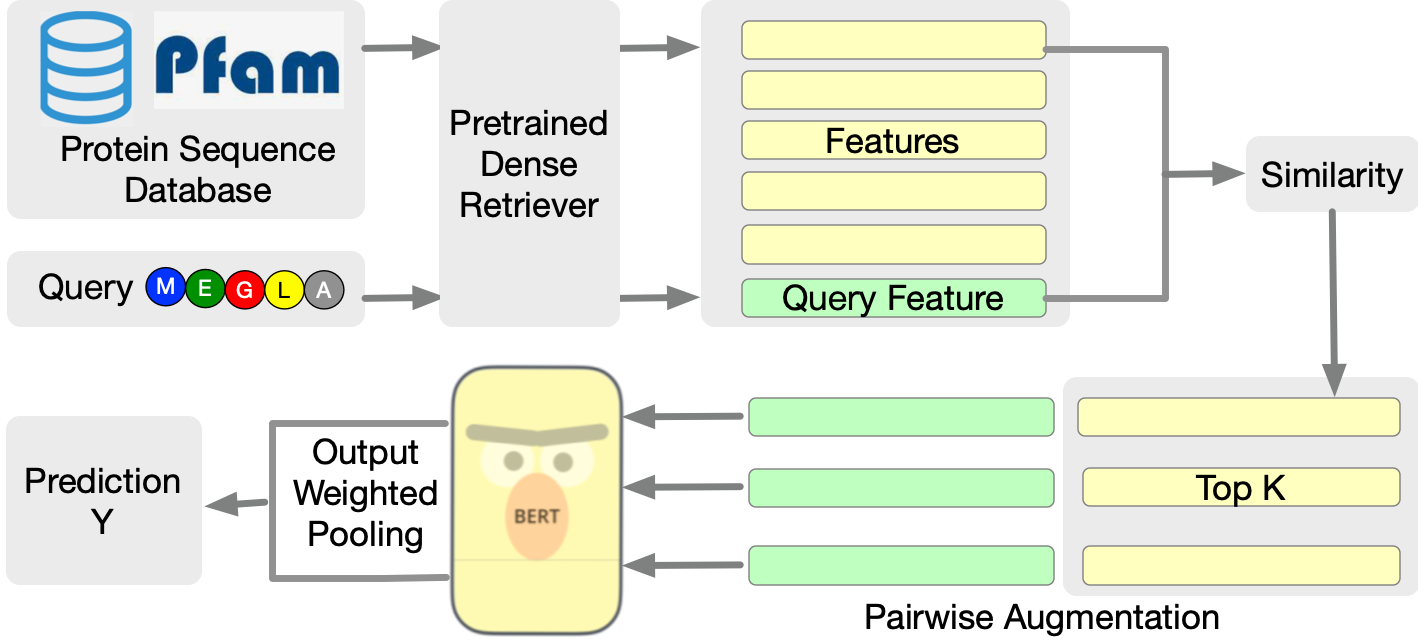
Figure 1: A brief overview of the proposed RSA protein encoding framework. Based on a query protein, RSA first retrieves related protein data from the database based on the top K similar features encoded by a pretrained retrieval model. Then we augment the query protein into pairs with each retrieved data and feed them into the protein model for protein tasks.
Comparison with MSA
The paper elucidates the conceptual alignment between MSA-based models and retrieval-augmented methods by demonstrating that MSA inherently performs retrieval-like augmentation. In RSA, retrieved protein sequences contribute similar evolutionary information to what MSA sequences provide, but without the burdensome alignment processes that typically slow down MSA methods. RSA achieves significantly faster computation, evidenced by a 373-fold speedup over conventional MSA methods on secondary structure prediction tasks.
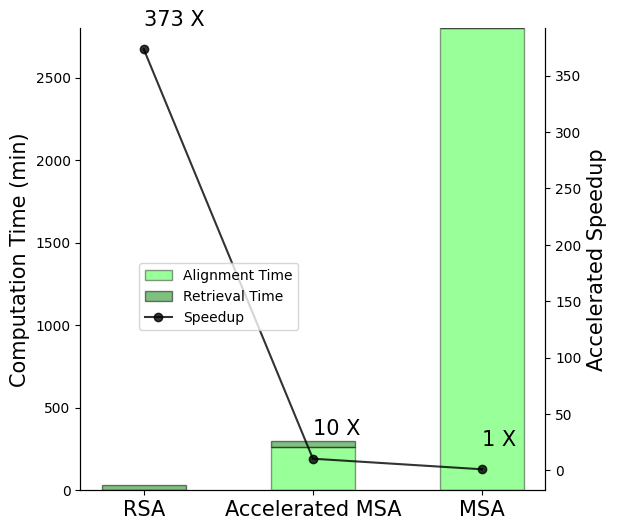
Figure 2: Illustration of speed up by RSA retrieval compared to MSA on secondary structure prediction dataset with 8678 sequences. Accelerated MSA refers to the MSA Transformer with MSA sequences retrieved by our RSA retriever.
Experimental Results
The RSA method shows robust improvements across a spectrum of protein prediction tasks, including secondary structure prediction, contact prediction, and homology prediction. The studies demonstrate RSA’s capability to outperform MSA Transformers by at least 5% on average, and the method excels especially in scenarios where domain adaptation is crucial, such as in predicting properties of de novo proteins.
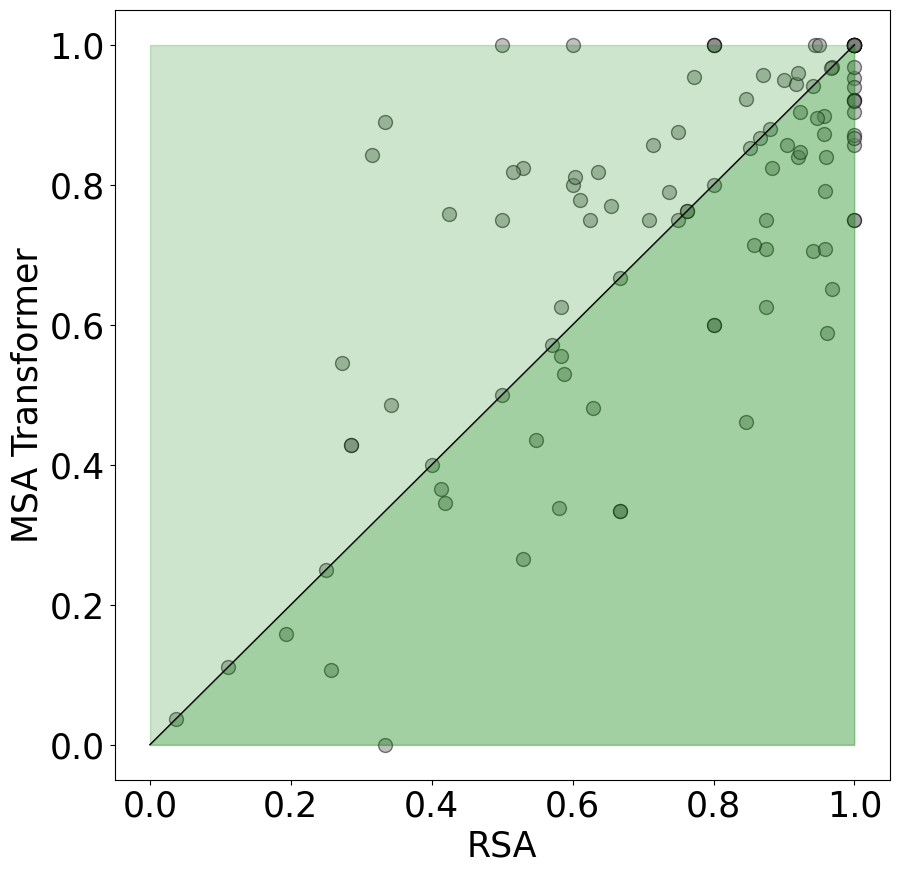
Figure 3: Contact Prediction of RSA and MSA Transformer on De Novo Proteins. We plot samples that RSA have better predictions under the diagonal line.
Moreover, the retriever used in RSA is shown to accurately identify homologous sequences with high precision and recall, supporting RSA's effectiveness in integrating meaningful protein representations.
Implications and Future Directions
The implications of RSA extend beyond mere performance improvements. By reducing computational demands, RSA opens avenues for accelerating drug discovery processes and refining protein engineering techniques without compromising predictive accuracy. Furthermore, its ability to adapt to novel protein domains positions RSA as a formidable tool in expanding our understanding of synthetic and modified proteins.
Future research could explore scaling RSA for broader protein databases and integrating three-dimensional folding tasks to complement sequence-based predictions. Additionally, exploring the potential of RSA in conjunction with other augmentation techniques could further enrich protein representation learning models.
Conclusion
The introduction of RSA marks a significant advancement in protein representation learning by circumventing traditional MSA limitations. It offers a streamlined, efficient, and potent approach that enhances both speed and accuracy, making it well-suited for rapid biological inference and discovery. The results affirm RSA’s role in propelling protein LLMs toward more adaptive and computationally feasible solutions.