- The paper presents a unified framework combining effective Hamiltonians, DFT, and many-body techniques to accurately describe both small and large polarons.
- It details computational strategies including supercell DFT corrections and machine-learned interatomic potentials to overcome self-interaction errors and capture lattice distortions.
- The work bridges theoretical models with experimental observables, validating ab initio predictions across materials like TiO₂, perovskites, and 2D systems.
Authoritative Summary of "Polarons from first principles" (2512.06176)
Introduction and Historical Models
"Polarons from first principles" provides a comprehensive review of recent advances in the ab initio theoretical study of polarons, focusing on unifying the diverse methodologies—ranging from effective Hamiltonians to many-body field-theoretic approaches—under a single conceptual and computational framework. The polaron, an emergent quasiparticle, results from strong electron–phonon interactions in crystals, leading to an electron or hole accompanied by a surrounding lattice distortion. The review highlights the evolution from early effective Hamiltonian models (Landau-Pekar, Fröhlich, Holstein, Su-Schrieffer-Heeger, Lee-Low-Pines, Feynman's variational method) to modern first-principles techniques rooted in density functional theory (DFT), Green’s function approaches, and ab initio diagrammatic Monte Carlo (DMC).
The Landau-Pekar and Fröhlich models describe large, delocalized polarons arising from weak electron-phonon coupling; conversely, the Holstein and related models capture the physics of small, localized polarons at strong coupling. The review details how the energetics and spatial extent of polarons interpolate smoothly across regimes; e.g., the Feynman path-integral approach achieves near-exact description at all coupling strengths, establishing benchmarks for controlled approximations and many-body computational schemes.
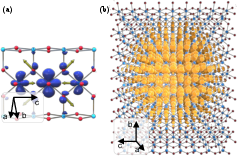
Figure 1: Visualization of small (a) and large (b) polarons in rutile TiO2 from ab initio calculations; isosurfaces show the polaronic wavefunction with local lattice distortion vectors.
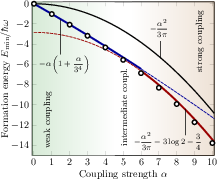
Figure 2: Fröhlich polaron formation energy as a function of coupling strength α; DMC data are shown alongside Landau-Pekar and Feynman results, delineating weak, intermediate, and strong-coupling regimes.
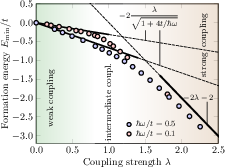
Figure 3: Holstein polaron formation energy in 1D as a function of electron-phonon coupling λ at varying adiabaticity; DMC results validate analytic weak/strong coupling limits.
DFT and Periodic Supercell Approaches to Polarons
The review provides a critical account of supercell-based DFT treatments of polarons and emphasizes their strengths and intrinsic limitations:
- Adiabatic and Classical Approximation: Ions are treated classically and the DFT total energy surface is explored to locate minima corresponding to polaronic configurations; electronic quantum fluctuations and nonadiabatic effects are neglected.
- Supercell Convergence: Finite-size effects, image charge interactions, and the need for large supercells for extended or weakly localized polarons are major challenges that require careful extrapolation or correction schemes to recover dilute polaron physics [Fig. 8].
- Self-Interaction Error (SIE): Standard semilocal functionals suffer from spurious self-Coulomb and exchange-correlation interactions, destabilizing polaron localization and underestimating formation energies. Hybrid functionals and Hubbard corrections provide partial mitigation but are parametrically sensitive and do not fully restore piecewise-linearity [Fig. 7].
Multiple self-interaction corrections are surveyed, including Koopmans-compliant and pSIC schemes, each with specific computational and conceptual tradeoffs. Post-processing approaches leveraging Janak’s theorem and density partitioning enable more robust extraction of polaron energetics in large systems.
Machine-learned interatomic potentials (MLIPs) are also discussed as a transformative advance, enabling long-timescale molecular dynamics simulations of polaron hopping and complex configurations, including multi-polaron and defect-mediated behavior, beyond the reach of brute-force DFT.
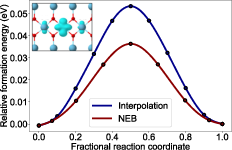
Figure 4: Polaronic migration/hopping barriers in rutile TiO2 calculated by NEB and interpolated paths; inset visualizes the charge density of the small electron polaron.
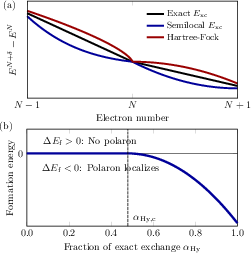
Figure 5: Piecewise linearity of total energy with electron number in exact DFT (black); semilocal and nonlocal functionals introduce curvature and SIE, affecting polaron energetics.
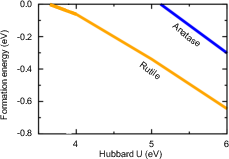
Figure 6: Polaron formation energy in DFT+U for anatase (hole) and rutile (electron) as U is varied; underscores sensitivity of polaron physics to correlation treatment.
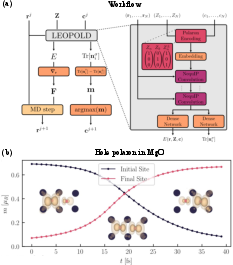
Figure 7: MLIP workflow for polaronic systems, combining site occupation tracking, energy/force prediction, and explicit polaron encoding (a), with time-resolved hopping event analysis (b).
Many-Body and Green's Function Approaches
The manuscript details several advanced ab initio many-body approaches for polarons. Fock-space Hamiltonians, parametrized entirely from DFT/DFPT (electron bands, phonon branches, matrix elements), serve as the foundation for canonical transformation (Lang-Firsov, Lee-Low-Pines, all-coupling variational), self-consistent Green’s function (Dyson/Hedin-Baym formalism), and (stochastic) diagrammatic Monte Carlo (DMC) [Figs. 12–15].
- Canonical transformation methods permit analytic or variational treatment of strong-coupling and small-polarons in complex materials, integrating with maximally-localized Wannier function analysis.
- Green’s function and cumulant expansion techniques provide momentum- and energy-resolved spectral functions, connecting directly with ARPES experiments and capturing non-quasiparticle structures (phonon sidebands, kinks) characteristic of polaronic systems [Figs. 10–11].
- Ab initio DMC enables parameter-free, all-coupling calculations of polaronic energies, spectral weights, and mass renormalizations, validating analytic and variational methods in both model and full first-principles settings. It recovers limiting behaviors of both Fröhlich and Holstein regimes and matches path-integral and Green's function techniques to high precision [Fig. 13].
An overarching field-theoretic formalism based on Hedin-Baym equations unifies the effective Hamiltonian and ab initio polaron equations, clarifies the structure of Fan-Migdal, Debye-Waller, and polaron self-energies, and provides an explicit route for including quantum, anharmonic, and screening effects. The nonlinear eigenvalue problem for the polaron—with self-consistent determination of lattice distortion and charge localization—emerges naturally in this rigorous framework [Fig. 14–15].
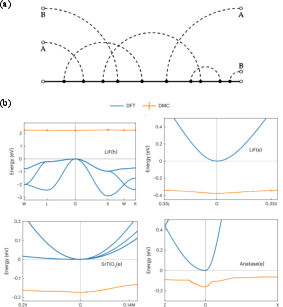
Figure 8: Sample Feynman diagrams used in DMC, with explicit electron and two-oscillator phonon lines; topology indexing for MC sampling.
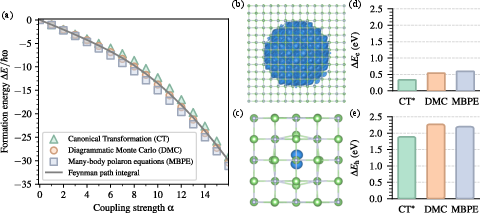
Figure 9: Formation energy of the Fröhlich polaron from canonical transformation (CT), DMC, and self-consistent Green's function (MBPE) approaches; data for LiF electron/hole polarons shown for each method.
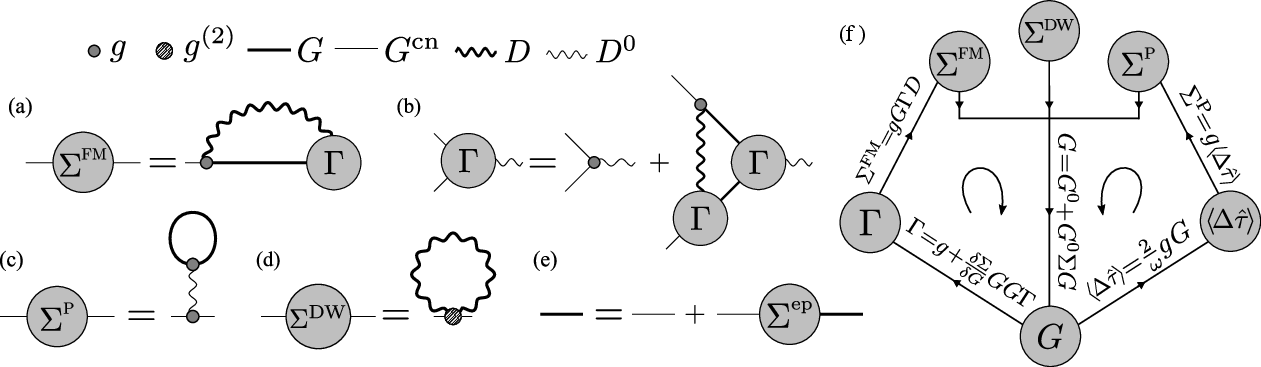
Figure 10: Diagrammatic representation of the various self-energies and Dyson equation in the self-consistent Green's function approach to ab initio polaron theory.
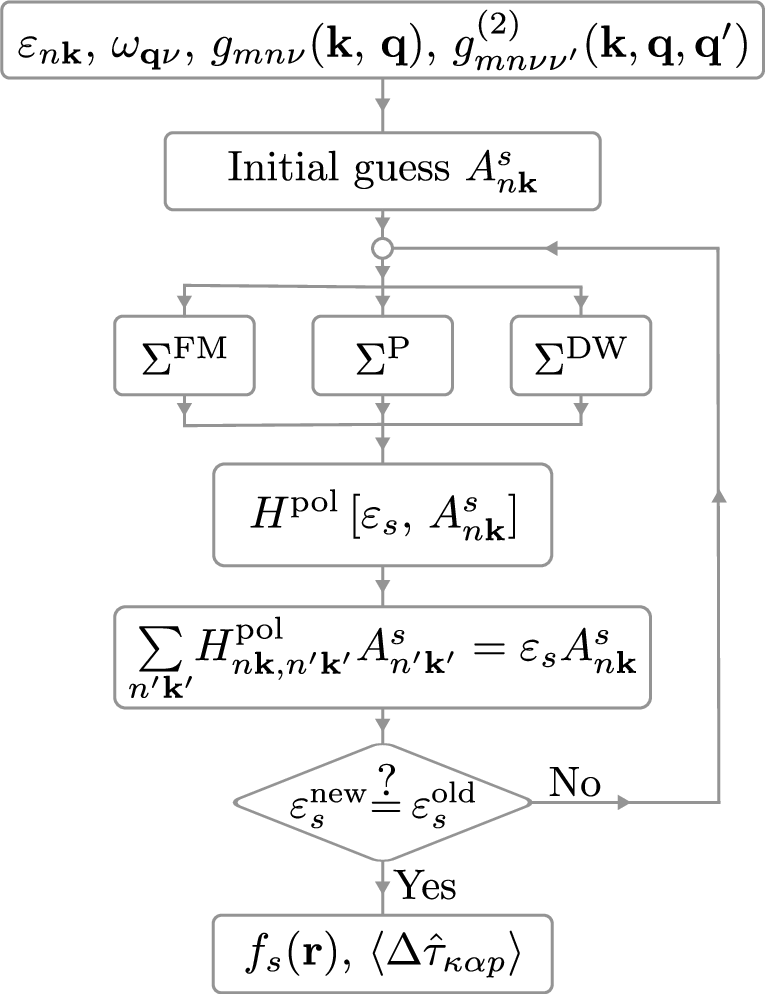
Figure 11: Flowchart of the self-consistent solution for Dyson orbitals and associated energies and displacements in the many-body polaron equations.
From Effective Theories to Real Materials: Connection and Applications
Interconnections between the approaches are mapped in a comprehensive “Jacob’s ladder” diagram, indicating tradeoffs between computational cost, atomistic detail, and quantum/classical lattice treatment [Fig. 16]. Key results include:
- GW, DFT, and many-body convergence: Polaron formation energies, spectral signatures, and dynamics obtained from Koopmans-compliant, GW, Green's function, and self-interaction-corrected DFT all converge in the dilute carrier limit for a variety of systems, validating the practical accuracy of ab initio polaron equations for large polarons.
- Role of anharmonicity and nonlinear coupling: For small polarons (and certain holes in materials like LiF), linear electron-phonon coupling and the harmonic approximation overestimate binding and localization, while large or adiabatic polarons are well described within these frameworks [Fig. 17].
Extensive applications are highlighted:
- Alkali halides: Both small and large polarons are captured with formation energies, spatial profiles, and migration barriers consistent across methods and in agreement with experiment; sensitivity to the treatment of self-interaction and parameter dependence is explicitly demonstrated [Fig. 20–22].
- Transition metal oxides (TMOs): Materials such as rutile and anatase TiO2, Fe2O3, and Li2O2 show complex polaron behavior, including coexistence of large and small polarons, strong dependence on correlation treatment (U, exchange fraction), and interface/surface effects; time-resolved STM experiments confirm polaron dynamics, and polaron migration barriers/transport can be directly linked with NEB and kinetic Monte Carlo analyses [Fig. 23].
- Halide perovskites (single and double): Both weakly and strongly localized polarons exist in hybrid and inorganic perovskites, with explicit connection to ultrafast optical experiments and the observed long carrier lifetimes and large Stokes shift [Fig. 24].
- Two-dimensional materials: Polaronic states in monolayer CoCl2 have been visualized, manipulated, and modeled with hybrid DFT and NEB; the unique critical condition for polaron formation in reduced dimensions is identified [Fig. 25].
- Transport: Large-polaron (bandlike) and small-polaron (hopping) transport are captured by Boltzmann equation and kinetic Monte Carlo frameworks, respectively. Advanced cumulant and DMC calculations recover activated and coherent regimes; however, the boundary between hopping and bandlike behavior remains an open question.
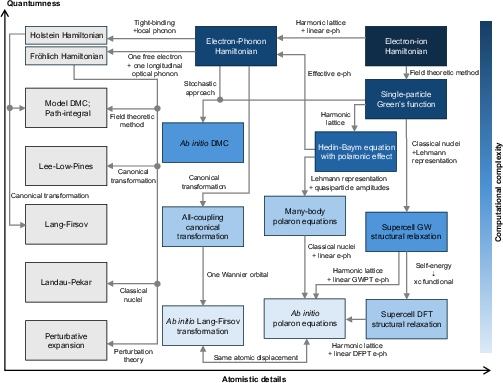
Figure 12: Mind map linking effective models, Fock-space ab initio, DFT-based, and Green's function methods for polarons, with computational scaling and lattice quantum/classical treatment axes.
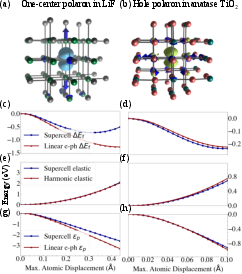
Figure 13: Direct comparison of supercell DFT and linear-response (polaron equation) approaches for small polaron energetics and lattice components in LiF and TiO2, illustrating the breakdown of the harmonic/linear-coupling approximation in certain cases.
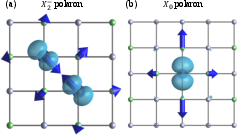
Figure 14: Visualization of two-center (X2−) and one-center (X0) small hole polaron states in LiF with atomic displacement vectors.
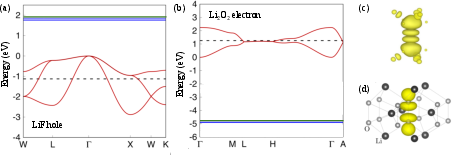
Figure 15: Formation energy comparison for X0 hole polaron in LiF; canonical transformation and ab initio equations yield mutually consistent results.
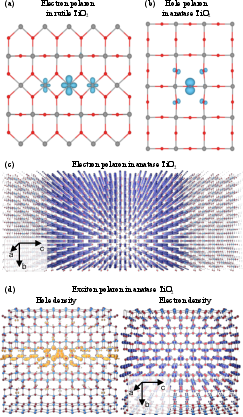
Figure 16: Small electron polaron (rutile TiO2), small hole polaron (anatase TiO2) from DFT, and large electron/exciton polarons visualized and analyzed in anatase TiO2.
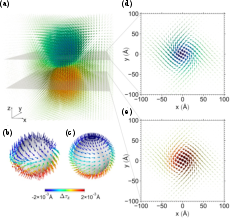
Figure 17: Non-trivial topological structure (Bloch points, meron vortices) in Ag displacement field of large electron polaron in double perovskite Cs2AgBiBr6.
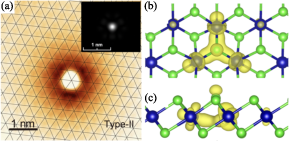
Figure 18: Experimental and calculated STM images and isosurface visualization of localized electron polaron in monolayer CoCl2.
Exciton Polarons and Extensions
- Exciton polarons and self-trapped excitons: Generalization of ab initio polaron equations to neutral excitations demonstrates consistent treatment of lattice-coupled photoexcited species in halide perovskites, α-SiO2, and other systems. New computational tools for the calculation of exciton-phonon couplings, utilization of the BSE kernel, and supercell vs reciprocal-space methods are detailed [Fig. 18–19].
- Topological and beyond-adiabatic phenomena: Topological textures in polaronic lattice distortions are identified in double perovskites. The theoretical framework incorporating nonadiabatic, quantum, and many-body effects is outlined, with explicit links to experimental observables (optical, ARPES, transport).
Open Problems, Contradictory Claims, and Prospectus
Notable and potentially contradictory claims include:
- For small alkali halide polarons, DFT/pSIC and hybrid functional supercell approaches may yield different qualitative ground states (one-center vs two-center), highlighting sensitivity to anharmonicity, functional choice, and lattice quantum effects.
- The existence and nature of electron/hole polarons in rutile vs anatase TiO2 depend critically on (and sometimes conflict across) the choice of U parameter, exchange fraction, SIE treatment, and computation of Koopmans compliance.
Implications and Future Outlook:
- The integration of DFT, many-body, and ML approaches ensures that predictive, parameter-free polaron calculations are approaching the level of quantitative confidence previously limited to mainstream condensed matter observables.
- Transport calculations unifying coherent and incoherent regimes, explicit inclusion of nonadiabatic/anharmonic effects, and systematic assessment of polaron–polaron and bipolaron formation will further expand the reach and reliability of these simulations.
- There is a clear path for the unification of Green's function and variational frameworks with real-time, finite-temperature, and strong-coupling extensions, as well as the benchmarking and validation needed to achieve consensus for application in emerging quantum materials science.
- The theoretical prospectus includes extending all-coupling approaches to exciton polarons, mapping collective effects with ML and DMC, and benchmarking various SIE/pSIC and GW-based frameworks to resolve the remaining discrepancies.
Conclusion
This review establishes a rigorous hierarchy and interconnected map between classical effective models, DFT-based approaches, Fock-space Hamiltonians, and advanced Green’s function and quantum Monte Carlo techniques for polarons and related excitations. The combination of analytic, numerical, and ML methodologies enables comprehensive exploration of polaronic phenomena across materials classes, dimensionalities, and coupling regimes. Current open questions—regarding exciton polaron unification, finite-temperature/correlation-driven transitions, bipolarons, and nonadiabatic dynamics—set the stage for vibrant further developments and essential connections with experiment and device innovation.

