- The paper presents a novel CAV strategy to localize protein motifs by leveraging PLM embeddings, achieving an F1 score over 0.85 on diverse motifs.
- It employs a linear logistic regression to distinguish motif from non-motif windows, yielding interpretable geometric representations in high-dimensional embedding space.
- The method efficiently detects multiple and cryptic motif instances, complementing traditional annotation tools for enhanced protein function analysis.
Automated Motif Localization in Protein LLM Embeddings via Concept Activation Vectors
Introduction
Protein motifs and domains are the modular determinants of protein function, defining catalytic sites, binding interfaces, and structural frameworks. Classical motif annotation pipelines dominated by profile-HMMs (e.g., Pfam, InterPro, PROSITE) yield extensive—and high-fidelity—motif databases, but are largely constrained to explicit sequence similarity and fail at reliable identification of motifs with high sequence divergence, remote homologs, or decoupled conservation patterns. Recent advances in protein LLMs (PLMs) present an opportunity for fundamentally novel approaches to motif discovery, leveraging geometric structure in high-dimensional embedding spaces learned from large-scale unsupervised sequence data.
In "Automated Protein Motif Localization using Concept Activation Vectors in Protein LLM Embedding Space" (2511.21614), the authors introduce an automated, interpretable framework for motif localization based on Concept Activation Vectors (CAVs)—a method adapted from neural network interpretability research—applied to PLM embeddings of protein sequence segments. Motifs are encoded as geometric directions, learned by linear separation of motif-containing and non-motif contexts in embedding space. The core hypothesis is that PLM representations allow motifs and functional patterns to be represented as linearly separable subspaces, enabling efficient, interpretable, and scalable motif annotation in arbitrary proteins.
Methodological Framework
The pipeline consists of two principal stages: (1) learning motif-specific CAVs from curated positive and negative subsequence windows, and (2) scoring and localization of motifs within novel protein sequences by projecting window embeddings onto CAVs.
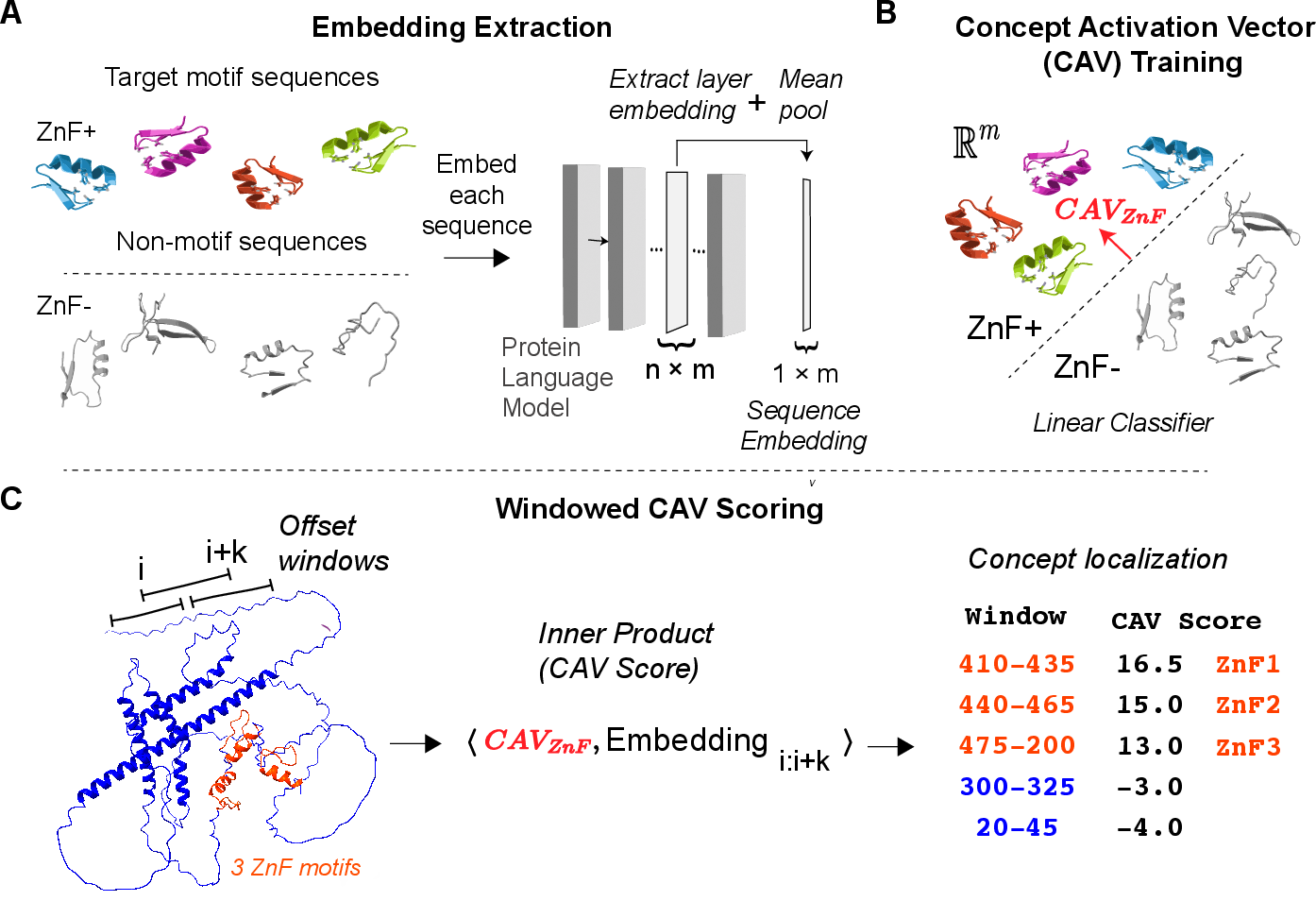
Figure 1: Schematic of the CAV-based motif localization pipeline—from extracting PLM embeddings of annotated and control windows (A), to linear CAV learning (B), to scoring candidate segments in query proteins (C).
Motif Concept Vector Construction
Positive sequence windows are constructed by centering on annotated motif occurrences with a context proportional to the median motif length. Negative examples are sampled from motif-excluding proteins. Each window is independently embedded using a PLM (ESM-C), with mean pooling employed to obtain a fixed embedding for each window. Linear logistic regression is trained to separate motif and non-motif windows. The normal vector of the resulting hyperplane is taken as the CAV, serving as a directional embedding for the motif concept.
A dictionary of CAVs is constructed, one per motif/family, capturing the geometric direction in embedding space most associated with that motif. This allows encoding of biochemical or structural properties as quantitative linear subspaces.
CAV-Based Localization and Scoring
For query proteins, overlapping windows are processed analogously, embedded, and each embedding is scored by the inner product with the target motif's CAV. This yields a position-wise score for motif-likeness. High-scoring segments are considered candidate motif localizations, with window stride and buffer parameters controlling coverage sensitivity.
Results and Analysis
PLM Layer Selection and Motif Signal
A series of analyses examined the propagation and locality of motif signal across PLM layers. The study identified a crucial trend: low-level layers exhibit broad, weak motif responses, while mid-late layers (notably ESM-C layer 26) concentrate strong, discriminative signal. Deepest layers show some reduction in motif-specific separability, suggestive of task-specific abstraction or compression.
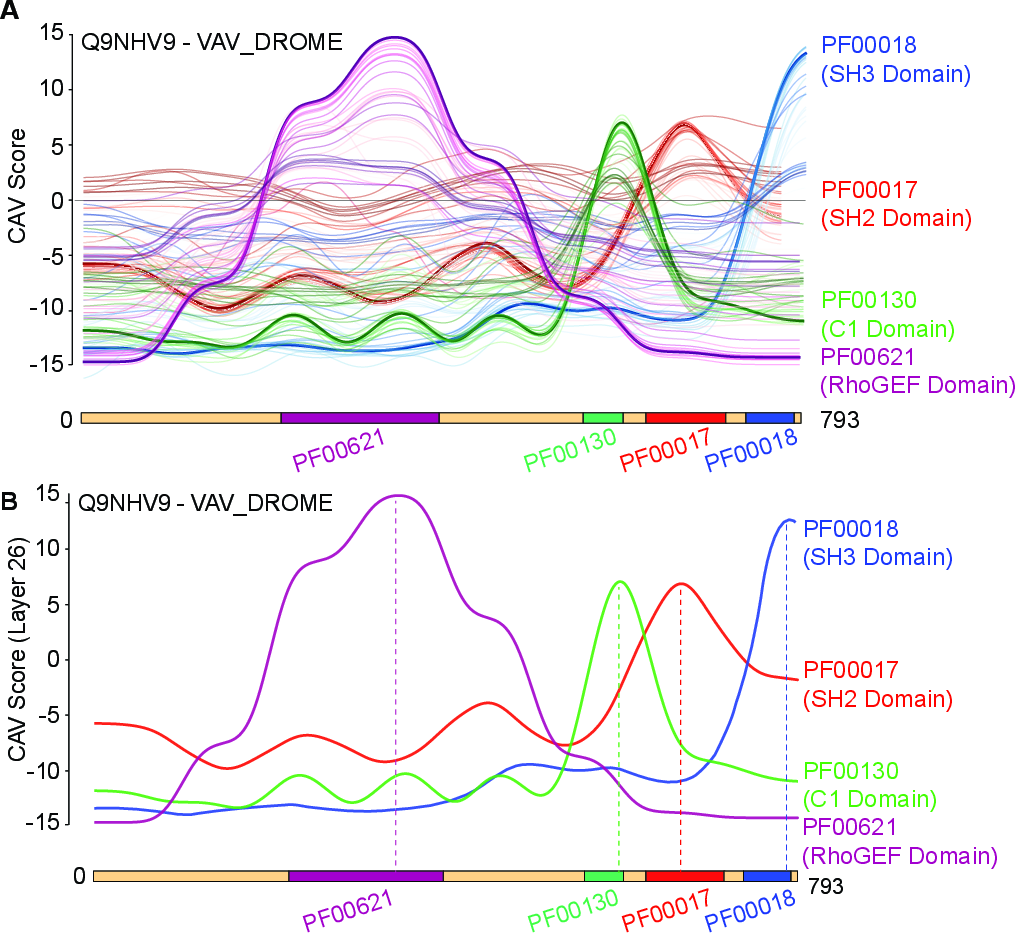
Figure 2: (A) Layer-wise CAV response profiles across ESM-C layers for motif localization in a representative protein. (B) CAV peaks map to empirically annotated domain intervals, supporting the hypothesis of localized motif encoding in PLM embedding space.
Multiple Motif Instances and Comparison to Deep Learning Annotation
The scoring mechanism naturally enables detection of multiple motif/domain occurrences within a protein (including situations not explicitly annotated in standard resources such as UniProt Features). The authors demonstrate localization to secondary/cryptic motifs—e.g., a second PF00076 count in SET1_SCHPO, detected as a sharp CAV peak but missed by standard annotations. Furthermore, CAV-based localization can recover motif instances overlooked by contemporary deep learning segmentation models (e.g., InterPro-N).
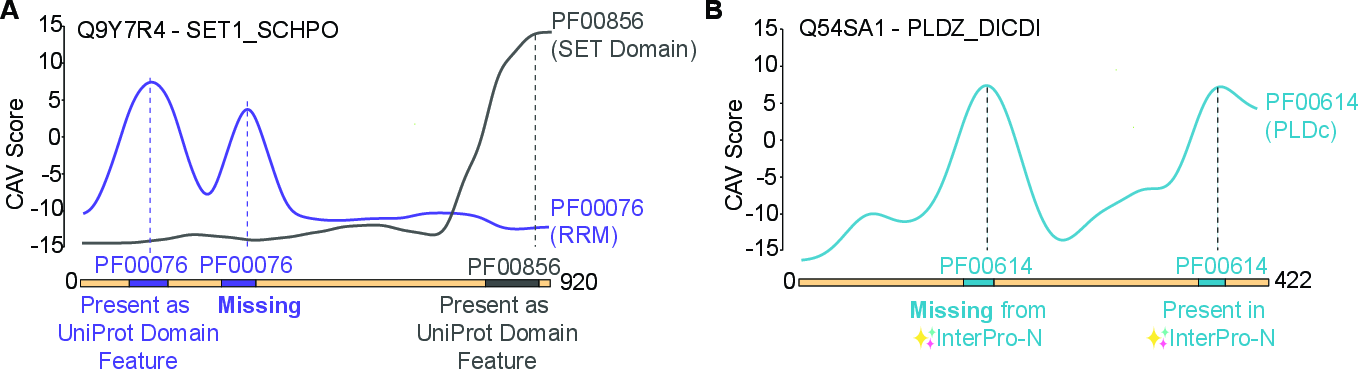
Figure 3: (A) CAV-based detection of multiple motif instances, including cryptic domains absent from reference annotations. (B) Demonstration of CAV outperforming an InterPro-N transformer model in uncovering a PLDc motif.
This finding highlights the method’s ability to complement and extend existing motif annotation tools, circumventing the biases and coverage limitations of alignment and segmentation-based models.
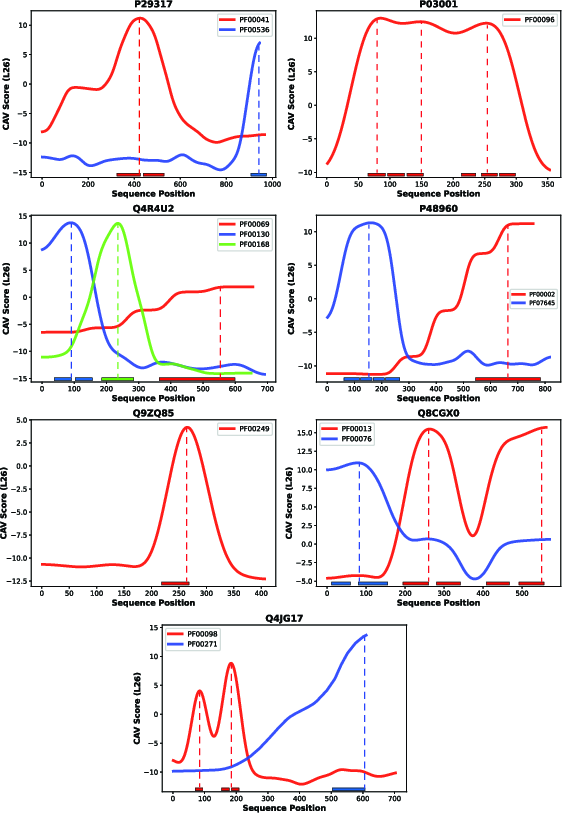
The approach was benchmarked across 69 motifs, spanning approximately 690 test proteins. On this diverse set, the method achieves an F1 score exceeding 0.85 for moderate overlap thresholds (10–30%), and retains reasonable fidelity (F1 0.80) up to a 60% overlap threshold. The precision and recall remain consistently high at lenient-to-moderate strictness, but the method displays a typical drop-off in boundary reproduction at stringent overlap cutoffs—reflecting both localization challenges and inherent ambiguity in curated motif boundaries.
The strong area-under-curve at practical overlap regimes supports the central claim that functional motifs are robustly encoded as linearly interpretable directions within large PLM embedding spaces.
Implications and Future Directions
The CAV framework for motif localization presents several significant theoretical and practical implications:
Conclusion
This study establishes that pretrained protein LLMs encode motifs and domains as geometrically coherent, linearly separable directions in embedding space. By leveraging directly constructed CAVs, motif localization is reframed as an efficiently solvable, interpretable geometric scoring problem. The approach attains high precision and recall on a diverse benchmark of Pfam motifs, accurately identifying motif-rich segments and multiple domain copies, including instances missed by canonical annotation resources and specialized deep learning models. Motif boundaries remain a challenge for precision, but the underlying representation is shown to be robust.
The CAV framework’s scalability, interpretability, and efficiency position it as a compelling foundation for next-generation protein annotation pipelines and open the door for theoretical advances in understanding the geometry of learned protein function spaces. Future work may explore dynamic motif discovery, multi-resolution scoring, and fusion with structural inference for holistic annotation systems.