Performance of Automatic Active Space Selection for Electronic Excitation Energies
Abstract: Computation of electronic spectra is one of the most important applications of methods capturing static electron correlation, including complete-active-space self-consistent field (CASSCF) and post-CASSCF theories. Performance of these techniques critically depends on the active space construction, both in terms of accuracy and computational effort. In this work we benchmark the performance of automatic active space construction, as implemented in the Active Space Finder software, for the computation of electronic excitation energies. The multi-step procedure constructs meaningful molecular orbitals and selects the most suitable active space based on information from more approximate correlated calculations. It aims to tackle a key difficulty in computing excitation energies with CASSCF: choosing active spaces that are balanced for several electronic states. The Active Space Finder is tested with several established data sets of small and medium-sized molecules and shows encouraging results. We evaluate multiple setting configurations and provide practical recommendations.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper is about teaching a computer program how to choose, by itself, the “right” pieces of a molecule to focus on when predicting the colors of light a molecule can absorb. That prediction is called an electronic excitation energy. The program, called Active Space Finder (ASF), helps set up a kind of quantum chemistry calculation (CASSCF) that needs a good “active space” to work well. Picking that active space by hand is tricky, slow, and sometimes subjective. The authors test how well ASF can pick these active spaces automatically and reliably.
What questions did the researchers ask?
They asked:
- Can a computer automatically choose good active spaces for molecules so that calculations of excited-state energies (the energy to excite electrons with light) are accurate and stable?
- Which settings of the ASF program work best?
- When does the automatic method struggle, and how can we fix those cases?
How did they do it?
Think of a molecule as a building with many rooms (orbitals). Electrons are like students who can move between rooms. An “active space” is the small set of rooms where the most important action happens during excitement (light absorption). Picking these rooms well makes the calculation both accurate and fast.
Here’s the simple version of their approach:
Step 1: Build a simple picture (Hartree–Fock)
They start with a basic, fast method (Hartree–Fock) to get an initial picture of the molecule’s orbitals. Sometimes they allow “symmetry breaking” (unrestricted HF), which lets the picture adapt better to complicated electron behavior.
Step 2: Pick a big enough “playground” (MP2 natural orbitals, with an optional QRO tweak)
They run a slightly more detailed method (MP2) to estimate how much each orbital is used (“occupation numbers”). From this, they pick an initial batch of orbitals likely to matter. Two flavors:
- Use these “natural orbitals” directly.
- Or rotate them into “QRO-like” orbitals (think: tidy up the rooms so they look more like standard shapes for excited states) without mixing in extra rooms.
Step 3: Quick test drives (DMRG-CASCI)
Before committing, they run a quick, low-accuracy test (DMRG-CASCI) on the initial set. You can imagine this like a short rehearsal to see which rooms are actually important when students (electrons) start moving around.
Step 4: Let the data choose the team (cumulant and entropy)
They analyze two simple-but-informative measures:
- Two-electron cumulant: tells which orbitals “talk” or correlate strongly with each other (which rooms are used together).
- One-orbital entropy: tells how “busy” an orbital is (how much it changes between on/off/half-filled).
They try several possible active spaces and pick the one whose least-busy member meets a target “busy-ness” (entropy) level. This balances “not too small” (misses important action) and “not too big” (too slow).
Special twist for excited states (averaging over states)
To predict excitation energies, you care about more than one state (ground and excited). They improve balance by:
- Averaging the “talking” (cumulant) and “busy-ness” (entropy) over the first two singlet states, so the chosen active space works fairly for both.
- In some setups, they also build orbitals using a triplet-state guess, which can give a nicer starting point for excited states.
After picking the active space, they run the main calculation (state-averaged CASSCF) and then add a correction step (NEVPT2) that mops up extra details the first step misses.
They tested all this on 32 small and medium molecules from well-known benchmark sets, using the same fair settings for everyone.
What did they find? Why does it matter?
Big picture: Automatic selection worked well most of the time, and best with certain settings.
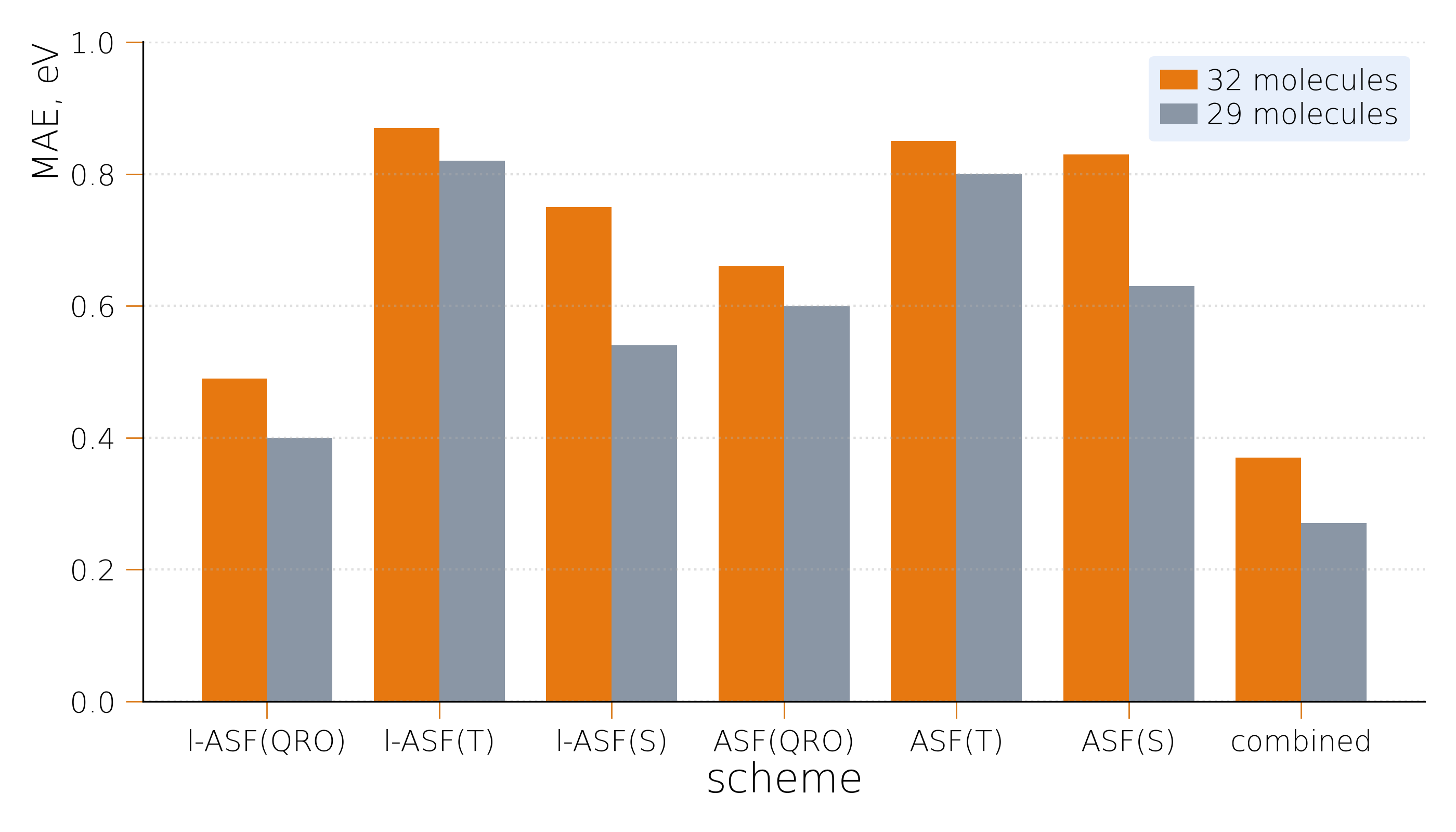
- Across all tested setups, the average error was under 1 eV (electron volt, a standard energy unit). The best setup had about 0.5 eV average error.
- The best-performing recipe was called l-ASF(QRO): it uses a slightly looser selection (lower entropy threshold) and rotates the starting orbitals into QRO-like shapes. With this, all calculations converged, and the average error was roughly 0.5 eV.
- Problems mainly happened in two situations: 1) Near-degenerate excited states: when two excited states are extremely close in energy, the method that averages over just two states can get confused about which state is which. Example: p‑benzoquinone. 2) Active space too small: if the automatically chosen space has too few orbitals, it can miss key behavior.
They also suggested a simple combined workflow: try the best setup (l-ASF(QRO)); if it produces a tiny active space (e.g., too few empty orbitals), switch to a singlet-based setup; if it’s still tiny, use the QRO version anyway but be cautious. This reduced errors further.
Why it matters: Picking the active space is the most frustrating, expertise-heavy step in these calculations. A tool that does it well automatically makes high-quality excited-state predictions more accessible, faster, and more consistent.
What does this mean going forward?
- Less guesswork: Researchers who aren’t specialists can still run solid excited-state calculations without hand-tuning orbitals.
- Faster screening: Useful for studying light-absorbing molecules in chemistry, materials, and photochemistry—think solar cells, sensors, LEDs, and drug photostability.
- Clear tips: If results look shaky, try:
- Averaging over more excited states (not just two).
- Allowing a slightly larger active space (lower the entropy threshold).
- Using QRO-like starting orbitals.
The software (ASF) is open-source and built on established tools, so the community can improve it further—especially for tricky cases with many close-lying excited states.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a focused list of what remains missing, uncertain, or unexplored, formulated to be concrete and actionable for future research.
- Generality across chemical space:
- Benchmark on broader and more diverse sets (e.g., QUESTDB subsets) including larger molecules, radicals, transition-metal complexes, heavier elements (with relativistic/ECP treatments), and excitation types beyond simple valence (charge‑transfer, Rydberg, double excitations).
- Assess robustness on systems with pronounced multireference character where MP2-based natural orbitals may be unreliable.
- Treatment of excited-state manifolds:
- Implement and benchmark the “union of individual active spaces” strategy head‑to‑head against the current “averaging” strategy (for same multiplicity) across many systems.
- Extend averaging to mix different spin multiplicities (S and T) with proper weighting; compare equal vs optimized weights and quantify impact on root-flipping and near-degeneracies.
- Develop automatic detection of near-degenerate manifolds at the DMRG-CASCI step and dynamic adjustment of the number of states included in averaging.
- Active-space size and selection criteria:
- Systematically study sensitivity to the entropy threshold beyond the two values tested (0.11, 0.14); devise an automatic, data-driven or stability-based threshold selection strategy.
- Quantify the relationship between minimum number of virtual orbitals in the active space (e.g., the “≥3 virtuals” heuristic) and accuracy, and validate or refine this heuristic across diverse excitation classes.
- Analyze how active space size distribution (e.g., (4,4) vs (8,9)) correlates with errors per molecule and excitation type; provide guidance for when to expand or contract spaces.
- Initial orbital choices:
- Evaluate alternatives to UHF‑MP2 natural orbitals (e.g., ROHF, QRO from ROHF, UNOs, KS-DFT natural orbitals, localized orbitals) for excited-state ASF; identify conditions under which each choice performs best.
- Investigate when QRO rotation improves performance and why (orbital character retention, reduced spin contamination, better state balance); provide diagnostics to decide when to apply QRO.
- DMRG-CASCI setup and cumulant analysis:
- Report and vary the DMRG bond dimension and truncation parameters used for cumulant/entropy estimates; quantify the effect of low-accuracy settings on active-space decisions and final excitation energies.
- Validate the stability and reliability of two-electron cumulant analysis versus mutual information/entanglement measures; identify cases where cumulant-based pairing may mislead selection.
- Explore orbital ordering and localization effects in DMRG (and in the ASF pipeline) on cumulant patterns and selected spaces.
- State-averaged vs state-specific workflows:
- Benchmark ASF for state-specific CASSCF (SS-CASSCF) and compare performance to SA-CASSCF; assess whether per-state tailored active spaces reduce misses and failures.
- Investigate automatic determination of SA weights (instead of fixed equal weights) based on preliminary state energies or entropic measures.
- Dynamic correlation treatment:
- Compare SC‑NEVPT2 with partially contracted NEVPT2, CASPT2 (with IPEA/shift), MR‑LCC, and other post‑CASSCF methods to quantify sensitivity of ASF-selected spaces to the dynamic correlation model.
- Assess susceptibility to intruder states with different post‑CASSCF methods and whether ASF choices mitigate or exacerbate intruders.
- Basis set dependence:
- Quantify basis-set sensitivity by benchmarking across diffuse/augmented sets (for Rydberg/CT states), core‑valence sets, and different zeta qualities; provide recommendations per excitation type.
- Investigate whether initial MP2 natural-orbital selection is more/less stable under basis changes than QRO or other orbital bases.
- Metrics, error analysis, and diagnostics:
- Provide more granular error analysis (per excitation type, per chemical class), beyond global MAE and >1 eV “miss” classification; include median, percentiles, and outlier characterization.
- Develop automatic sanity checks (e.g., root-tracking, state overlap monitoring, state character analysis) to flag likely failures prior to production SA‑CASSCF/NEVPT2.
- Correlate convergence behavior (iterations, root swaps) with ASF parameters and initial orbital choices to meet the stated requirement of “good SCF/CASSCF convergence.”
- Scope limitations of current study:
- Extend beyond vertical energies: assess adiabatic excitations with excited-state geometry optimization and vibronic structure (Franck–Condon/Huang–Rhys), and evaluate whether ASF-selected spaces remain balanced along excited-state PES.
- Include properties beyond energies (oscillator strengths, transition dipoles, spin–orbit couplings) to ensure orbital choices preserve state character relevant to spectroscopy.
- Reproducibility and transparency:
- Release the excited-state ASF code updates and document all parameter values (DMRG bond dimension, initial-space size limits, occupation thresholds, density-fitting settings) to enable full reproducibility.
- Provide standardized workflows and input templates, plus open datasets with per-molecule active-space decisions, to facilitate community benchmarking.
- Comparative benchmarking:
- Perform head‑to‑head comparisons against existing automatic selection methods (autoCAS/QICA(S), ABC family, ASS1ST, AVAS/SPADE, ranking/scoring, ML/data-driven approaches), using consistent protocols and metrics.
- Scalability and performance:
- Characterize computational cost and scaling (time, memory) of ASF steps (MP2 natural orbitals, DMRG‑CASCI, cumulant/entropy analysis) as a function of system size and initial-space width; propose optimizations for high-throughput use.
- Open methodological questions:
- Investigate principled ways to combine cumulant analysis with entropy thresholds (e.g., multi-objective optimization) rather than selecting by “closest to target” lowest entropy.
- Explore adaptive workflows that iterate ASF steps (e.g., expand/reduce active space or change initial orbitals) in response to diagnostics, while preserving a priori and reproducible character.
Practical Applications
Immediate Applications
Below are specific, deployable use cases that can be adopted now, along with sector links, potential tools/workflows, and feasibility notes.
- Automated active-space selection in excited-state workflows (software, materials, pharma)
- What to do: Integrate ASF’s l-ASF(QRO) protocol (lowered entropy threshold + QRO rotation) into state-averaged CASSCF + SC-NEVPT2 pipelines for vertical excitation energies to reduce manual curation.
- Tools/workflows: PySCF + ASF + Block2; orchestration with AiiDA/FireWorks/Snakemake on HPC; standardized job templates using def2-TZVPD and state-averaged over two singlets.
- Value: Lowers expert time and boosts reproducibility; achieves ~0.5–0.8 eV MAE on small/medium organics as benchmarked in the paper.
- Assumptions/dependencies: Open-source ASF and PySCF/Block2 stack; performance shown for small/medium organic molecules, vertical excitations, def2-TZVPD, SC-NEVPT2; accuracy sensitive to near-degenerate states and small active spaces.
- High-throughput screening of organic chromophores where TD-DFT struggles (materials, coatings, consumer goods, healthcare)
- What to do: Use ASF-driven SA-CASSCF/NEVPT2 to triage candidates for dyes, UV absorbers, photoresists, fluorescent probes when single-reference methods are unreliable.
- Sectors: OLED emitters/dopants (electronics), solar dyes (energy), UV stabilizers (polymers/coatings), phototoxicity flags in drug design (healthcare).
- Tools/workflows: Library enumeration → ASF active spaces → SA-CASSCF/NEVPT2 vertical excitations → rank and filter.
- Assumptions/dependencies: Limited to vertical excitations; triplets and S–T gaps require explicit setup; cost scales with active space size; include “small active space” guardrail (e.g., require ≥3 virtual orbitals).
- Diagnostic triage for challenging excited-state cases (R&D, method support)
- What to do: Use ASF’s low-bond-dimension DMRG-CASCI as a quick pre-check to identify near-degeneracy and root-swapping risk; trigger automatic expansion of state averaging or active-space size.
- Tools/workflows: Implement the paper’s combined protocol (start with l-ASF(QRO), fallback to l-ASF(S), warn on tiny active spaces) with standardized flags/alerts in CI pipelines.
- Assumptions/dependencies: Requires capturing at least the lowest 4 electronic states in the pre-check to detect near-degeneracy; results depend on UHF and MP2 natural orbital quality.
- Rapid rescue of TD-DFT failures (industrial computational chemistry support)
- What to do: When TD-DFT delivers inconsistent excitations, automatically switch to ASF-selected SA-CASSCF/NEVPT2 for the same geometry.
- Sectors: Specialty chemicals, pigments/inks, battery electrolyte additives, photoinitiators.
- Tools/workflows: Conditional workflow nodes that promote systems based on multi-reference indicators (spin contamination, multi-config diagnostics) to ASF multireference path.
- Assumptions/dependencies: Requires pre-screening criteria for multi-reference character; computational budget must accommodate CASSCF/NEVPT2.
- Education and reproducible method training (academia)
- What to do: Use ASF in graduate teaching labs to demonstrate active-space selection, entropy/cumulant diagnostics, and state-averaging pitfalls.
- Tools/workflows: Notebooks in PySCF/ASF; assignments comparing default vs lowered entropy threshold, and QRO vs natural orbitals.
- Assumptions/dependencies: Requires access to modest HPC; focuses on small/medium molecules.
- Pre-processing for quantum computing and CASCI/DMRG studies (software, quantum technologies)
- What to do: Use ASF’s a priori active spaces for CASCI/DMRG or variational quantum eigensolver (VQE) benchmarks in excited states.
- Tools/workflows: Export ASF-selected active spaces/orbitals to quantum workflows; prepare symmetry-consistent orbital subsets.
- Assumptions/dependencies: Current results emphasize organic molecules; triplet and multi-multiplicity averaging require care; quantum hardware scale may limit active space size.
- Internal best-practice playbooks and templates (industry, academia)
- What to do: Publish an internal SOP adopting l-ASF(QRO) as default, the combined protocol for small-active-space alerts, and a “more-states” fallback for suspected near-degeneracy.
- Tools/workflows: Checklists: minimum number of virtual orbitals in active space, entropy-threshold ranges, inclusion of >2 states for molecules like p-benzoquinone/DMABN.
- Assumptions/dependencies: Tailor thresholds to portfolio chemistry; maintain versioned ASF/PySCF environments for reproducibility.
Long-Term Applications
These opportunities require further method development, scaling, or broader validation before routine deployment.
- Industrial-grade black-box excited-state service with confidence metrics (software, materials, healthcare)
- Vision: SaaS/on-prem solution that automates ASF-driven active-space selection, state-averaging, and post-CASSCF correlation, with uncertainty flags (e.g., near-degeneracy, small-space, spin-state issues).
- Product features: GUI, REST API, automated choice of number of averaged states, adaptive entropy thresholds, state-tracking/root-following, and standardized reports.
- Dependencies: Robust handling of larger molecules, transition-metal systems, multiple spin manifolds; improved root-tracking and automatic state inclusion.
- Expansion to adiabatic excitations, vibronic spectra, and dynamics (materials, photochemistry, photobiology)
- Vision: ASF-guided on-the-fly active-space updates along excited-state geometries for adiabatic energies, vibronic couplings, and nonadiabatic dynamics.
- Tools/workflows: Coupling ASF with trajectory surface hopping/MCTDH and geometry optimization; active-space continuity enforcement.
- Dependencies: Algorithms for stable active-space transport along PES; efficient multi-state averaging; cost mitigation for repeated CASSCF/NEVPT2.
- Automated multi-multiplicity averaging and excited-state root tracking (software/methods)
- Vision: Generalized averaging over spin multiplicities and robust root-following to avoid state mixing and root-swapping across scans.
- Tools/workflows: Spin-aware cumulant/entropy averaging with weights, diagnostics for state identity conservation, adaptive re-averaging.
- Dependencies: New theory/software features (currently averaging in ASF implemented for same multiplicity); careful design of weighting schemes.
- Scaling to transition-metal complexes and strongly correlated materials (catalysis, energy, quantum materials)
- Vision: Apply ASF principles with localized/fragment orbital pipelines (e.g., AVAS/SPADE hybrids) and DMRG to larger, metal-containing systems.
- Tools/workflows: Fragment-aware initial spaces, spin-state ladders, automated ligand-field-aware selection.
- Dependencies: Validation on broad metal benchmarks; handling dense manifolds and near-degeneracies; computational cost control.
- Integrated design loops for optoelectronic materials and photocatalysts (materials, energy)
- Vision: Close the loop between materials informatics (generative models), ASF-based multireference validation, and experimental design of chromophores/OLED emitters/photocatalysts.
- Tools/workflows: Active learning that flags multi-reference regimes and prioritizes ASF routes; property targets (absorption maxima, S–T gaps).
- Dependencies: Harmonized datasets at scale (beyond Thiel/QUESTDB), surrogate models for triaging, cost-aware scheduling.
- Community standards and regulatory guidance for multireference excited-state evidence (policy, industry consortia)
- Vision: Best-practice standards (FAIR data, reproducible pipelines, reporting checklists) for regulatory submissions involving colorants, UV filters, and photostability claims.
- Tools/workflows: Public benchmark suites, reference implementations, validated protocols for automatic active-space selection.
- Dependencies: Multi-stakeholder consensus; broader validation across chemistries, including solvents and finite-temperature effects.
- Interoperability and data schemas for active spaces (software ecosystems)
- Vision: Cross-code plugins and a standardized schema for storing/transferring ASF-selected active spaces and diagnostics among PySCF, ORCA, Q-Chem, Molpro, BAGEL, etc.
- Tools/workflows: Common orbital/active-space exchange formats; provenance capture; containerized environments.
- Dependencies: Vendor/community collaboration; careful handling of orbital phases/symmetries.
- Quantum-classical hybrid pipelines for near-term devices (quantum technologies)
- Vision: ASF as a front-end to map chemically relevant active spaces onto quantum processors for excited states (e.g., ADAPT-VQE, subspace expansion).
- Tools/workflows: Hardware-aware truncations, error mitigation tailored to excited states, state-averaged ansätze.
- Dependencies: Device scale and noise; excited-state VQE robustness; consistent orbital bases across classical/quantum stacks.
- Crowd-sourced benchmark expansion and challenges (academia, consortia)
- Vision: Expand beyond existing datasets (Thiel, QUESTDB) with communal benchmarks for excited-state multireference problems and automatic active-space selection challenges.
- Tools/workflows: Leaderboards, shared CI pipelines, result reproducibility badges.
- Dependencies: Data licensing, quality control, sustained hosting/funding.
Cross-cutting assumptions and dependencies (affecting feasibility across applications)
- Scope: Current benchmarks focus on small/medium organic molecules, vertical excitations, and SC-NEVPT2 with def2-TZVPD; transfer to metals/large systems needs validation.
- Method choices: Best performance reported for l-ASF(QRO); results may degrade for near-degenerate manifolds unless more states are averaged; small active spaces often underperform.
- Software stack: ASF (open source) depends on PySCF and Block2; production use requires managed environments and HPC resources.
- Cost and scaling: CASSCF/NEVPT2 costs grow rapidly with active-space size; DMRG mitigates but introduces its own parameters (bond dimension).
- Robustness: Intruder-state issues are reduced with NEVPT2 but method-specific artifacts can appear; reliable root tracking and spin/multiplicity averaging are active development areas.
Glossary
- ABC family: Adaptive basis sets designed to optimize correlated wave functions in quantum chemistry. "Recent developments along these lines have lead to adaptive basis sets for correlated wave functions (ABC family) \cite{Bao2018, Bao2019}"
- Active Space Finder (ASF): Open-source software that automatically constructs and selects active spaces for multireference electronic structure methods. "This work employs the Active Space Finder (ASF) package,\cite{ASF_github}"
- Active space: A selected subset of molecular orbitals included explicitly in multireference calculations to capture essential correlation effects. "The active space size affects both accuracy and time-to-solution: in a straightforward implementation, CASSCF complexity scales exponentially with active space size."
- Adiabatic excitations: Electronic transitions considering nuclear relaxation to the equilibrium geometry of the excited state. "Additional challenges arise if one is interested not only in vertical excitations, but in adiabatic ones and in vibronic structure."
- ASS1ST: An automatic active space selection scheme based on first-order perturbation theory. "active space selection based on 1st order perturbation theory (ASS1ST) \cite{Khedkar2019, Khedkar2020} scheme."
- Atomic valence active spaces (AVAS): A projector-based technique to construct active spaces focusing on atomic valence characteristics. "Examples are atomic valence active spaces (AVAS) \cite{Sayfutyarova2017, Claudino2019, Kolodzeiski2023, Lei2021}"
- Auxiliary basis sets: Additional basis sets used to approximate electron repulsion integral computations efficiently (e.g., in density fitting). "using the corresponding auxiliary basis sets\cite{Weigend2008}"
- Block2: A density matrix renormalization group (DMRG) solver/library interfaced with PySCF. "Block2\cite{Zhai2021} was employed via its PySCF interface for DMRG calculations called by the ASF."
- CASCI: Complete active space configuration interaction; a multireference method using a fixed set of orbitals without orbital optimization. "such as complete active space configuration interaction (CASCI)."
- CASSCF: Complete active-space self-consistent field; a multireference method optimizing orbitals and configuration interaction within an active space. "Computation of electronic spectra is one of the most important applications of methods capturing static electron correlation, including complete-active-space self-consistent field (CASSCF) and post-CASSCF theories."
- def2-TZVPD: A triple-zeta quality basis set augmented with diffuse functions for improved description of excited states and anions. "The triple-zeta quality basis set with diffuse functions def2-TZVPD \cite{Weigend2005, Rappoport2010} was used for all calculations."
- Density fitting (Resolution-of-identity): An integral approximation technique to accelerate electron repulsion integral evaluation. "Resolution-of-identity (or density fitting) \cite{VAHTRAS1993} was applied to accelerate electron repulsion integral evaluation"
- Density matrix renormalization group (DMRG): A variational tensor-network method for solving high-dimensional quantum many-body problems, used here to approximate CASCI. "The central component of our active space finding procedure is a density matrix renormalization group (DMRG)\cite{chan2011density} calculation performed with low-accuracy settings."
- Dipole moment-based scheme: An approach to active space selection that utilizes dipole-moment information (for excited states). "Of particular interest for the scope of this work are the methods extended to treat excited states: autoCAS, ABC2 and the dipole moment-based scheme \cite{Kaufold2023}."
- Dynamic correlation energy: Correlation effects due to rapid electron motions beyond static correlation, typically recovered by post-CASSCF methods. "As a post-CASSCF method to calculate the dynamic part of the correlation energy we chose second-order n-electron valence state perturbation theory, NEVPT2 \cite{angeli2001introduction}."
- Fock matrix: The one-electron operator matrix in mean-field theories (e.g., HF), whose diagonalization yields canonical orbitals. "the respective Fock matrix sub-blocks are projected to the initial active space constructed from MP2 natural orbitals prior to diagonalization."
- Hartree-Fock (HF): A mean-field electronic structure method providing reference orbitals and densities for correlated calculations. "Spin in the HF (SCF) calculation:"
- Intruder states: Spurious near-degenerate or energetically problematic states that destabilize perturbative correlation treatments. "Additionally, some perturbation theories for dynamic correlation are prone to intruder states."
- Matrix product state bond dimension: The parameter controlling the accuracy/complexity of the tensor-network (MPS) representation in DMRG. "This step is carried out as a DMRG-CASCI calculation with a low matrix product state bond dimension."
- Møller–Plesset perturbation theory (MP2): Second-order perturbation theory on the Hartree–Fock reference, often used to generate natural orbitals and densities. "natural orbitals of an orbital-unrelaxed second-order M\o{}ller-Plesset perturbation theory (MP2) density matrix for the ground state."
- Mutual information: An information-theoretic measure of correlation between orbitals derived from entropies. "In contrast to mutual information calculated from two-orbital entropies,\cite{Stein2016} which include contributions of reduced density matrix elements of up to fourth order,\cite{Boguslawski2015}"
- Natural orbitals: Orbitals obtained by diagonalizing the one-particle density matrix, often used to identify correlated subspaces. "All procedures used in the present work employ natural orbitals of an orbital-unrelaxed second-order M\o{}ller-Plesset perturbation theory (MP2) density matrix for the ground state."
- NEVPT2: N-electron valence state perturbation theory (second order), a robust multireference perturbation method. "As a post-CASSCF method to calculate the dynamic part of the correlation energy we chose second-order n-electron valence state perturbation theory, NEVPT2 \cite{angeli2001introduction}."
- Occupation number threshold: A criterion based on orbital occupation numbers used to select or truncate an initial active orbital set. "With the help of an occupation number threshold, an initial set of orbitals is selected, and further orbitals are discarded if their number exceeds an upper limit."
- One-orbital density: The probability distribution over local occupancy states for a single spatial orbital, used for averaging and entropy. "The averaged one-orbital density for each spatial orbital is calculated using the respective one-orbital density of each state :"
- One-orbital entropy: An entropic measure of single-orbital correlation/occupation fluctuations used to guide active space selection. "In order to select one of these active spaces, the one-orbital entropy is used as an auxiliary criterion.\cite{Boguslawski2012,Boguslawski2015}"
- Projector/fragment-based techniques: Methods that use projections onto fragments or atomic subspaces to define active spaces. "Alternative approaches to active space selection exploit projector/fragment-based techniques."
- QICAS: Quantum-information-assisted CAS optimization method using information measures to improve active space choices. "and the quantum-information-assisted CAS optimization (QICAS)\cite{Ding2023} method."
- Quasi-degenerate (excited states): Excited states with very small energy separations that complicate state averaging and optimization. "Error analysis has revealed two main sources of poor performance: the presence of quasi-degenerate excited states not considered in the calculation"
- Quasi-restricted orbitals (QROs): Orbitals resembling a restricted open-shell HF solution, used here via a transformation within the initial space. "initial space in a procedure that is analogous to quasi-restricted orbitals (QROs).\cite{Neese2006}"
- Reduced density matrix: A lower-order density matrix (e.g., one- or two-electron) encoding subsystem correlations. "which include contributions of reduced density matrix elements of up to fourth order,\cite{Boguslawski2015}"
- Restricted Hartree-Fock (RHF): HF variant enforcing paired spins in closed-shell systems. "However, it is also possible to carry out a restricted Hartree-Fock (RHF) calculation manually and to use it to proceed further with the ASF software."
- Restricted open-shell Hartree-Fock (ROHF): HF method with unpaired electrons treated in a restricted formalism. "even though it is represented by orbitals resembling a restricted open-shell Hartree-Fock (ROHF) solution."
- Root-swapping: The inadvertent change of the targeted electronic state during iterative optimization. "This can lead to root-swapping in the CASSCF procedure."
- SC-NEVPT2: Strongly contracted NEVPT2; a computationally efficient contraction scheme for NEVPT2. "We utilize the strongly-contracted scheme for NEVPT2 (SC-NEVPT2) \cite{ANGELI2001297}"
- Singlet: A spin multiplicity where total spin S=0; often refers to ground or excited electronic states with paired electrons. "However, it is also possible to carry out a restricted Hartree-Fock (RHF) calculation manually and to use it to proceed further with the ASF software." (e.g., "even if the system has singlet multiplicity.")
- SPADE: Automatic partition of orbital spaces based on singular-value decomposition. "automatic partition of orbital spaces based on singular-value decomposition (SPADE) \cite{Claudino2019, Kolodzeiski2023}"
- Spin multiplicity: The number 2S+1 indicating total spin state (e.g., singlet, triplet), crucial for orbitals and state averaging. "Often, it will be advantageous to perform the Hartree-Fock calculation, and the subsequent MP2 calculation (if applicable), for the state with the highest spin multiplicity."
- State-averaged CASSCF: CASSCF formulation optimizing orbitals averaged over multiple electronic states. "This permits the approach to be applied in a more general context to large active spaces, for which calculations may be expensive. It also makes the scheme suitable for methods that do not optimize orbitals, such as complete active space configuration interaction (CASCI)." (used throughout as "state-averaged CASSCF")
- State-specific CASSCF: CASSCF optimization targeted to a single electronic state. "which is needed to compute electronic excitations either by state-averaged or state-specific CASSCF."
- Static (strong) electron correlation: Correlation arising from near-degenerate configurations not captured by single-reference methods. "The complete active-space self-consistent-field (CASSCF) method and multireference approaches based on it are standard tools to capture static (strong) electron correlation \cite{roos1987complete}."
- Two-electron cumulant: The connected part of the two-electron reduced density matrix isolating true correlation beyond mean-field products. "An important design goal of the ASF was to identify correlation partner orbitals. This is accomplished through an automatic analysis of the two-electron cumulant."
- Two-electron density matrix: The second-order reduced density matrix encoding pairwise electron correlations. "only the two-electron density matrix is required to compute the two-electron cumulant."
- Unrestricted Hartree-Fock (UHF): HF variant allowing different spatial orbitals for alpha/beta spins, enabling symmetry breaking. "The fully automatic mode of the ASF always employs the spin-unrestricted Hartree-Fock (UHF) method."
- Unrestricted natural orbitals (UNOs): Natural orbitals derived from unrestricted HF, often used to identify minimal active spaces. "Pulay and co-workers demonstrated the utility of unrestricted natural orbitals (UNOs) for the selection of minimal active spaces.\cite{pulay1988uhf,pulay2015unos}"
- Vertical electronic excitations: Excitation energies computed at fixed ground-state geometry without nuclear relaxation. "This work focuses on vertical excitation energies between the ground and the first excited state"
- Vibronic structure: Coupling and spectral features arising from simultaneous electronic and vibrational transitions. "Additional challenges arise if one is interested not only in vertical excitations, but in adiabatic ones and in vibronic structure."
Collections
Sign up for free to add this paper to one or more collections.