- The paper introduces a novel inverse formulation that models molecular conformations as continuous probability distributions rather than discrete states.
- The method employs statistical measures like KL divergence and MMD within a Wasserstein gradient flow framework to optimize cryo-EM image reconstruction.
- Synthetic experiments validate the approach by robustly capturing structural heterogeneity in biomolecules, with potential integration with traditional MAP techniques.
Cryo-EM as a Stochastic Inverse Problem
Introduction
Cryo-electron microscopy (Cryo-EM) is a formidable technology for imaging biomolecules at high resolutions. However, its capability is curtailed by the structural heterogeneity of biomolecules, which poses a significant challenge in 3D reconstruction. Conventional methodologies assume a finite set of states, thus failing to capture the continuous variability of molecular structures. This paper proposes a novel approach by recasting Cryo-EM reconstruction as a stochastic inverse problem over probability measures. Instead of representing structures as discrete states, this formulation models molecular conformations as a continuous distribution subject to a random forward operator. The problem is thus posed as the minimization of a statistical discrepancy between observed and simulated image distributions. This approach is validated on synthetic data of proteins, showing that it effectively captures the continuous spectrum of states.
Methodology
The paper formulates the Cryo-EM reconstruction as a stochastic inverse problem represented in the space of probability measures. The observed 2D cryo-EM images are modeled as the push-forward of an unknown distribution over molecular conformations via a random forward operator. The authors propose solving the reconstruction problem through optimization over statistical distances, such as the Kullback-Leibler (KL) divergence and Maximum Mean Discrepancy (MMD), within a framework of Wasserstein gradient flow. This optimization is performed over probability measure spaceutilizing particles to represent and evolve conformational ensembles.
Variational Approach
The variational problem is defined to minimize the statistical discrepancy between the empirical distribution of observed cryo-EM images and the distribution of simulated images derived from a candidate structural distribution. This discrepancy is quantified using statistical measures, including the KL divergence and the MMD. The paper emphasizes using a gradient flow in the Wasserstein space to navigate the optimization landscape, which is numerically solved through a particle-based method.
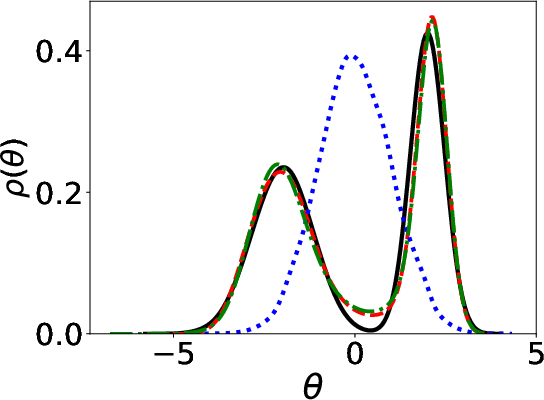
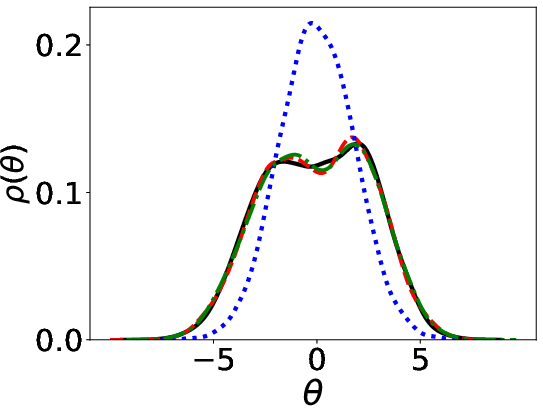
Figure 1: Parameter distributions (initial, estimated, ground truth) using Energy distance and KL divergence.
Numerical Validation
The proposed framework is tested using synthetic examples, including a model of heterogeneously structured nanoclusters and a realistic protein model. These tests demonstrate the method's proficiency in reconstructing continuous distributions of structural states, capturing the molecular conformations that contribute to the biological functionality of proteins.
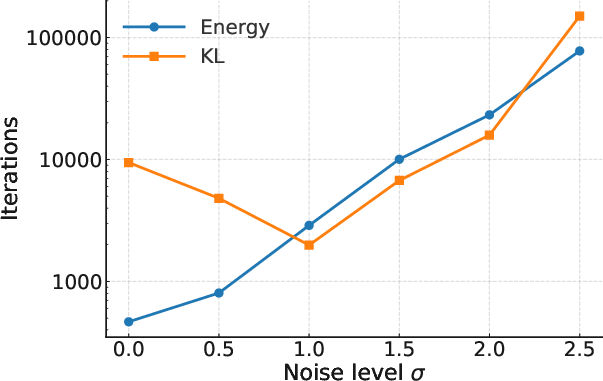
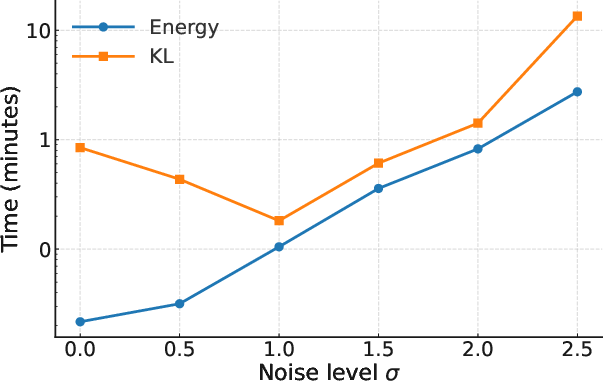
Figure 2: Iterations needed to reach W2<0.2.
In modeling protein conformations using normal modes, the results illustrate the method's potential to recover distributions that reflect real physical variability within the observed structures. The study concludes that the optimization framework allows for a robust interpretation of cryo-EM data without dependency on discrete state assumptions, thereby offering a novel paradigm for approaching structural heterogeneity in biomolecules.
Integration with Existing Frameworks
The authors draw connections to conventional Maximum A Posteriori (MAP) estimation methods, illustrating how the proposed stochastic inverse problem framework aligns with and extends these established techniques. The analysis reveals how conventional methods can be interpreted as part of a discretize-then-optimize (DTO) strategy, whereas the novel methodology employs an optimize-then-discretize (OTD) approach. The research provides a comparative analysis of these paradigms and establishes conditions where DTO methods converge towards the solution of the continuous problem, thereby shedding light on the potential for integrating new methodological insights with traditional frameworks.
Conclusion
The exploration of cryo-EM as a stochastic inverse issue using optimal transport theory paves the way for more versatile modeling of structural heterogeneity in biomolecules. As opposed to traditional discrete-state modeling, this framework acknowledges the inherent variability of molecular conformations. The proposed methodology surpasses existing approaches by producing high-fidelity reconstructions of continuous structural landscapes and broadening the applicability of cryo-EM for elucidating biological processes. Its prospect in other domains involving stochastic inverse problems is promising, potentially leading to significant advances in structural biology and beyond.
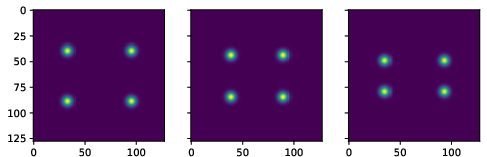
Figure 3: True data (noiseless images).

