- The paper introduces ML-based scoring functions using both experimental and computer-generated datasets to predict protein–ligand binding affinities.
- It details the use of fully-connected neural networks with tailored configurations, achieving moderate correlation coefficients (Rp ≈ 0.60) in predictive tests.
- The study demonstrates that per-target scoring functions may enhance application-specific modeling despite challenges with vertical test accuracy on unseen proteins.
Machine Learning Scoring Functions for Drug Discovery
Introduction
Scoring functions (SFs) are critical tools in computational drug discovery, used to predict the binding affinities between protein--ligand complexes. The paper investigates ML approaches to SF development, focusing on artificial neural networks and contrasting the performance of these methods using experimental and computer-generated data (2212.03202). ML SFs are posited to leverage vast structural databases to predict binding efficiency, potentially enhancing virtual screening processes and reducing drug discovery timelines.
Methodology
Data Sources
Two distinct databases were utilized: an experimental database with 2408 crystallographic protein--ligand complexes and a computer-generated database comprising 28,200 complexes. The experimental dataset was curated to include hydrogen atoms to enrich the data quality, while the computer-generated dataset was created using the CCDC Gold docking engine interfaced with MOE software (2212.03202).
Neural Network Configuration
The employed model is a fully-connected neural network (FCNN) with variable layer depth and width. Optimal network configurations differ by dataset size, with the experimental data requiring a simpler network due to its limited volume (Nl×Nh=2×20), whereas the more extensive computer-generated data benefitted from a deeper network (Nl×Nh=4×40).
Results
Horizontal Test Comparisons
Horizontal tests showed similar predictive results for both experimental and computer-generated datasets, suggesting the viability of using computer-generated structures for drug discovery.
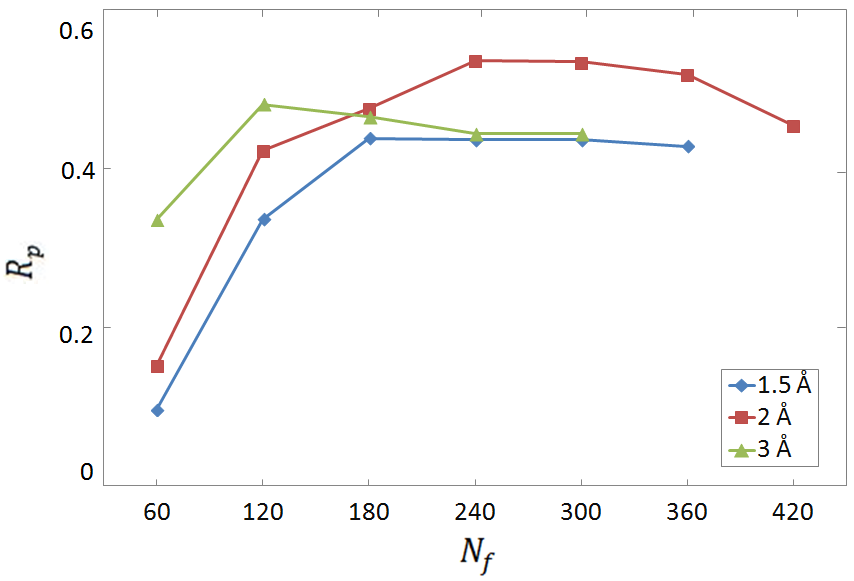
Figure 1: Pearson correlation coefficient Rp for the predictions of binding affinity provided by the FCNN SF, as a function of the number of descriptors Nf.
It was noted that larger datasets provided higher prediction accuracy, evidenced by moderately strong correlation values for binding affinity predictions (Rp≈0.60 for the computer-generated dataset) (2212.03202).
Vertical Tests
Vertical tests, which excluded proteins from the training set that appeared in the test set, revealed decreased accuracy, highlighting the importance of protein diversity in training datasets. The FCNN SF performed with moderate correlation (Rp≈0.4) for unseen proteins, showing a need for refined training methods.
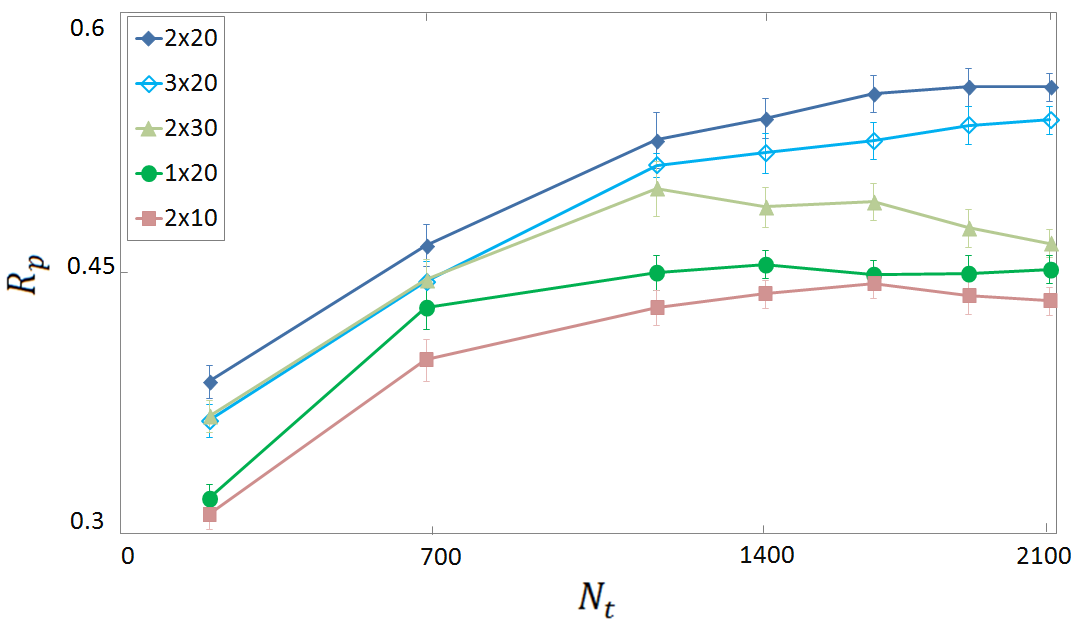
Figure 2: Rp score for binding affinity prediction in the vertical test as a function of the size Nt of the training set.
Per-Target Scoring Functions
The study explored per-target scoring functions using computer-generated datasets exclusively for specific proteins, showing increased correlation with larger datasets. This approach yielded encouraging results, indicating per-target SFs' potential in application-specific modeling.
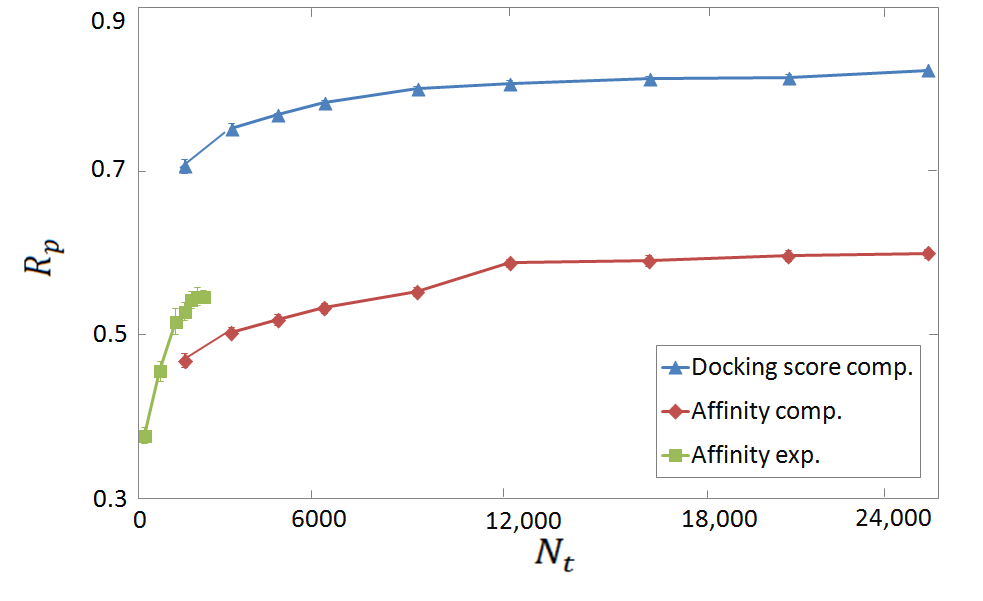
Figure 3: Rp scores for binding affinity predictions from per-target FCNN SF's for selected proteins.
Discussion
The research underscores the utility of computer-generated structures, matching experimental datasets in reliability and providing scalability due to easier data generation. While horizontal test results are promising, the discrepancy in vertical testing calls for more robust methodologies. The per-target SF approach represents a practical direction, utilizing computer-generated data for specific proteins, which may overcome generalized SF limitations.
Conclusion
This study presents a meaningful comparison between experimental and computational data usage in ML-based drug discovery, contributing valuable insights into SF development. Future work should focus on optimizing descriptor choice, exploring additional molecular interactions, and improving training protocols to enhance vertical test reliability. Broadening atomic species consideration will further refine predictions, aiding in SF accuracy and computational drug discovery efficiency (2212.03202).
This markdown essay provides a comprehensive summary of the paper's content, highlighting methodologies, results, and implications for both experimental and computational data in the field of machine learning-driven drug discovery. The visual figures underscore key data points, enhancing the readability and impact of the essay.