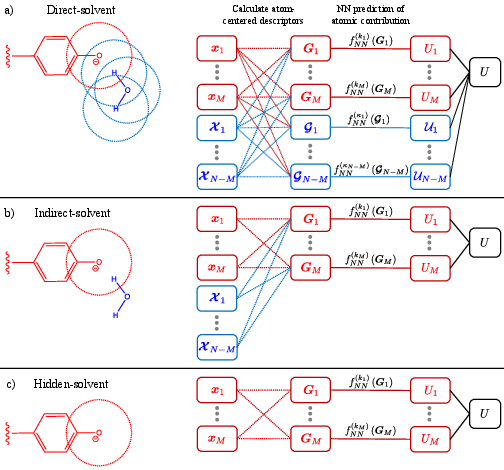
- The paper introduces a hierarchy of atom-centered neural network models (direct, indirect, hidden-solvent) to efficiently predict excited-state energy gaps in complex environments.
- The indirect-solvent model achieves sub-0.05 eV RMSE with 2000 training points, accurately reproducing key spectral features in both linear and two-dimensional measurements.
- The approach reduces computation costs by over two orders of magnitude, enabling enhanced sampling and integration with advanced quantum dynamical methods.
Machine Learning Acceleration of Multidimensional Optical Spectroscopy in Complex Environments
Introduction
The simulation of spectroscopic properties of chromophores in the condensed phase remains an outstanding computational challenge due to the burden of repeating high-level electronic structure calculations across extensive ensembles of nuclear configurations. The paper "Exploiting machine learning to efficiently predict multidimensional optical spectra in complex environments" (2005.09776) presents a structured approach for leveraging machine-learned models to predict excited-state energy gaps rapidly, thereby enabling efficient computation of both linear and multidimensional optical spectra in diverse solvated environments.
The authors introduce a hierarchy of atom-centered neural network (NN) approaches that differ in their treatment of the chromophore environment:
This structured hierarchy allows systematic exploration of the tradeoff between physical fidelity, model complexity, and data efficiency.
Data Requirements and Accuracy Characterization
Studies were performed on (a) the anionic photoactive yellow protein chromophore (pCT−) in water (strong, site-specific H-bonding), (b) Nile red in water (intermediate chromophore-solvent interactions), and (c) Nile red in benzene (weak interactions). The models were trained on subsets ranging from 500 to 8000 high-level vertical excitation energies, and their predictive RMSE (validation set) was assessed.
In the strong-coupling regime (pCT− in water), with increasing training set size, the more complex indirect- and direct-solvent models ultimately outperform the simplified hidden-solvent approach. Indirect-solvent achieves sub-0.05 eV RMSE with as few as 2000 training points, notably lower than the intrinsic energy gap fluctuation scale.
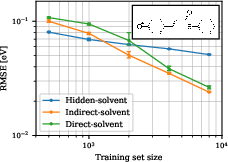
Figure 2: Learning curves for pCT− in water illustrate the crossover in model performance versus training set size.
Convergence of the ML-predicted energy gaps to the ab-initio target set directly translates to high-fidelity optical spectra:
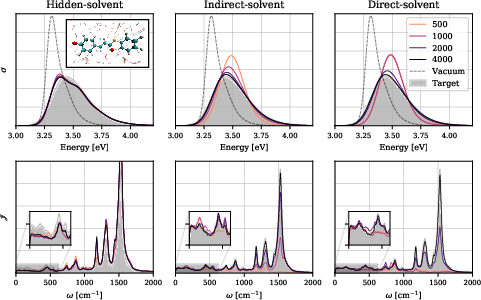
Figure 3: Absorption spectra and corresponding spectral densities for pCT− in water; only indirect- and direct-solvent models reach graphical fidelity.
The hidden-solvent approach systematically fails to reproduce low-frequency solvent-coupled spectral features and underestimates stochastic spectral diffusion, leading to incorrect vibronic structure, as evident in both linear and two-dimensional electronic spectra.
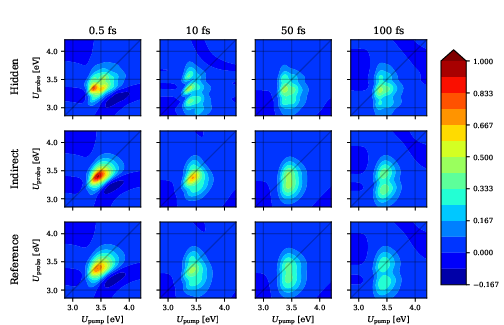
Figure 4: 2DES for pCT− in water; only the indirect-solvent model captures correct timescale-dependent features (Stokes shift, spectral diffusion) at long delays.
Dependence on Chromophore-Solvent Interaction Strength
The transferability of the above conclusions is established by comparing ML-model performance across the environments:
- In weakly-coupled systems (Nile red/benzene), the hidden-solvent model outperforms more complex alternatives at small and moderate training set sizes (≤4000), delivering high-quality linear and 2D spectra.
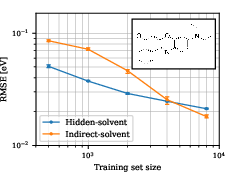
Figure 5: For Nile red/benzene, hidden-solvent models outperform indirect-solvent until large training sets are used.
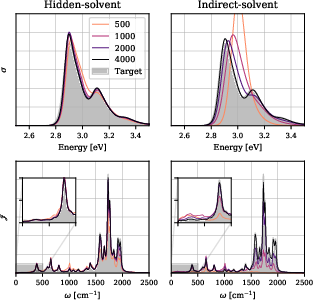
Figure 6: Convergence of absorption spectrum and spectral densities for Nile red/benzene as a function of model and training set size.
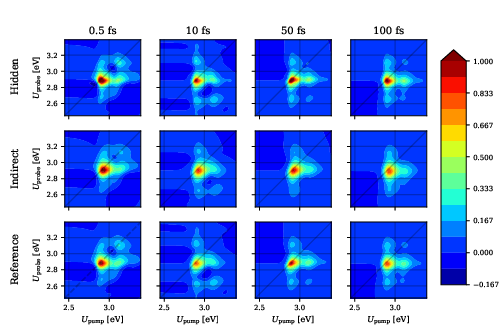
Figure 7: 2DES for Nile red/benzene recovered by both models, though indirect-solvent requires more data.
- In moderately-coupled systems (Nile red/water), indirect-solvent models are eventually necessary for accurate reproduction of spectrum and spectral diffusion when provided sufficient training data. Hidden-solvent models consistently exaggerate high-frequency chromophore vibronic structure.
These results underscore that locality of electronic excitations and chromophore-solvent coupling strength fundamentally determine the most efficient ML representation. For strong site-specific interactions, explicit local description of solvent atoms by indirect-solvent models is essential for capturing reorganization energy and stochastic bath fluctuations.
Implications and Future Applications
The computational impact is substantial: For a system requiring >30,000 energy gap calculations (each 8 hours), training on only 2000 reference points and using an ML surrogate reduces the total cost by over two orders of magnitude (savings of 240,000 GPU hours). Such efficiency gains open the door to broader and more accurate statistical sampling, higher electronic structure levels, and advanced dynamical treatments of excited state phenomena.
Theoretically, this work clarifies that physical model reduction (e.g., restricting descriptor cutoff, environment atom selection) must be tuned to system-specific electronic localization and environmental coupling. This recommendation extends to diverse quantum/classical embeddings, including QM/MM, multichromophoric assemblies, and spatially heterogeneous systems.
Looking forward, further integration with semiclassical and quantum dynamical methods will enable accurate on-the-fly simulations of nonlinear spectroscopies, excited state lifetimes, energy transfer, and photophysics in biomolecular and materials systems. The flexibility to localize the ML representation based on known excitation spatial character, or to adapt the cutoff to select specific bath degrees of freedom, positions these models for broader applicability beyond optical spectroscopy, e.g., to electron transfer or charge transport.
Conclusion
This work demonstrates that physically-motivated ML models, exploiting chromophore locality and systematically encoding solvation effects, achieve quantitative reproduction of both linear and multidimensional optical spectra with dramatically reduced electronic structure sampling. The choice of environmental treatment—hidden-, indirect-, or direct-solvent—should be guided by a priori estimates of chromophore-solvent coupling and available data, with indirect-solvent models ensuring accurate spectroscopic observables in strongly interacting regimes. This framework provides a robust, data-efficient protocol for the simulation of complex photoactive systems and will enable future combination with advanced quantum dynamical schemes for comprehensive investigation of condensate-phase excitonics and photochemistry.