- The paper introduces computational frameworks combining physics-based simulations and machine learning to design IDPs with controlled conformational ensembles.
- It demonstrates methods to optimize binding affinities and phase separation behaviors using coarse-grained models and deep-learning predictions.
- The work highlights actionable strategies for engineering IDRs for therapeutic and biomaterials applications through multi-objective optimization.
Computational Design of Intrinsically Disordered Proteins
Overview and Biological Significance of IDRs
Intrinsically disordered regions (IDRs) are prevalent in the human proteome, with approximately 70% of proteins containing at least one disordered stretch and 5% being fully disordered. Unlike globular proteins, IDRs lack stable secondary or tertiary structure, resulting in highly dynamic conformational ensembles that are sensitive to solution conditions, post-translational modifications, molecular crowding, and interactions with other biomolecules. This structural heterogeneity underpins their multifunctionality, enabling context-dependent roles in cellular signaling, molecular recognition, and phase separation. The versatility of IDRs has motivated efforts to engineer them for technological applications, including therapeutic targeting, biosensor design, and the development of stimuli-responsive biomaterials.

Figure 1: Technological applications of IDRs inspired by biological functions, including membrane receptor binding, scaffolding, chaperone activity, and formation of biomolecular condensates.
The functional design of IDRs requires a detailed understanding of the relationship between amino acid sequence and conformational ensemble. Key sequence features such as charge fraction, charge segregation, and aromatic residue patterning are conserved and modulate compaction, biological function, and cellular localization. Physics-inspired metrics (e.g., κ, sequence charge/hydropathy decoration) have enabled proteome-wide analyses to identify features associated with specific functions, which can be rationally introduced into synthetic sequences.
Molecular simulations, ranging from all-atom explicit solvent models to coarse-grained and residue-level approaches, have been instrumental in characterizing sequence-ensemble relationships. The computational efficiency of coarse-grained models (e.g., Martini, CALVADOS, Mpipi-GG) has facilitated large-scale exploration of IDR conformational space. Alchemical trajectory reweighting and Monte Carlo sampling in sequence space allow for rapid evaluation of sequence variants, enabling the design of IDRs with targeted compaction, contact maps, and environmental responsiveness.

Figure 2: Simulation-based approach for designing IDRs, illustrating force field resolutions, alchemical transformations, and optimization of contact maps.
Machine learning (ML) models trained on simulation data have emerged as efficient surrogates for predicting ensemble-averaged properties and generating conformational ensembles. Diffusion-based generative models and sequence-based approaches further expand the design space, though their accuracy is contingent on the quality of underlying physics-based models and training data. Notably, alignment-free representations and evolutionary information are increasingly leveraged for de novo design, but limitations remain in modeling environmental effects and secondary structure.
Design Strategies for Binding and Molecular Recognition
IDR-mediated binding encompasses a spectrum from folding-upon-binding to fuzzy complexes with persistent disorder. Computational design strategies address both the generation of IDRs with targeted secondary structure propensities and the engineering of high-affinity binders for IDRs. Physics-based approaches, such as ABSINTH simulations combined with genetic algorithms, enable the design of IDRs matching specific helicity profiles under compositional constraints.
For binder design, knowledge-based methods (Rosetta) and generative diffusion models (RFdiffusion) are used to create pockets accommodating IDRs in various conformations. These approaches are complemented by deep-learning models (AlphaFold2, ProteinMPNN) for backbone and sequence refinement. When the bound conformation is unknown, simultaneous sampling of IDR and binder conformations allows for adaptive complex formation. Sequence-based pipelines utilizing protein LLMs facilitate the design of short peptides with high specificity, even for targets with unknown structure, and have demonstrated efficacy in targeted protein degradation.
Recent advances include sequence-based prediction of two-chain interaction maps using residue-level force field parameters, enabling rational design of binders for fully disordered complexes. These methods provide efficient tools for exploring the vast landscape of IDR-mediated interactions.
Engineering Phase Separation and Selective Partitioning
IDRs drive the formation of biomolecular condensates via multivalent interactions, resulting in dynamic assemblies with distinct physicochemical properties. Early engineering efforts focused on elastin-like polypeptides (ELPs) and resilin-like polypeptides, elucidating rules linking sequence composition to phase behavior (LCST/UCST). Computational methods, including ABSINTH simulations and genetic algorithms, have expanded the accessible sequence space and enabled the design of IDRs with tailored phase separation (PS) propensities.
Systematic quantification of saturation concentration (csat) has facilitated residue-level ranking of PS contributions and informed coarse-grained modeling. Automated sequence optimization using genetic algorithms and fitness functions based on phase concentrations has enabled the design of multiphasic condensates with selective partitioning.
Conformational compaction, contact maps, and second virial coefficients serve as proxies for PS propensity, though their predictive power is limited by the complexity of intermolecular interactions. Sequence-based prediction of interaction maps and ML models trained on simulation data (with active learning) allow for large-scale design and optimization of condensate properties, balancing stability and dynamics.
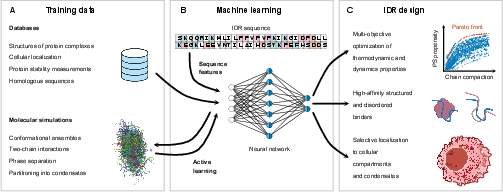
Figure 3: Role of machine learning in de novo design of IDRs, highlighting training on diverse data sources, active learning, and multi-objective optimization.
Selective partitioning into subcellular compartments is critical for therapeutic efficacy. Statistical analyses and deep-learning models trained on compartmentalization annotations enable the design of IDRs with targeted localization. The interface of biomolecular condensates, characterized by electric potentials and pH gradients, can be engineered for catalytic activity and therapeutic intervention. Optimization pipelines integrating simulations and ML surrogate models have successfully designed amphiphilic peptides that localize to condensate interfaces, validated across multiple scaffold systems.
Limitations and Future Directions
Despite significant progress, current molecular models often neglect transient secondary structure, ion-specific effects, and temperature-dependent hydrophobic interactions. Non-globular protein models struggle to reproduce experimental observables, and generative models inherit limitations from their training data. Deep-learning models trained on heterogeneous datasets may suffer from limited transferability and benchmarking challenges.
Future developments should focus on improving the accuracy of conformational ensemble and phase behavior predictions, incorporating environmental effects, and developing alignment-free representations for rapidly evolving IDR sequences. The integration of coarse-grained simulations, ML surrogate models, and active learning offers a robust framework for multi-objective optimization in complex systems. Systematic computational investigations will enhance understanding of sequence-biophysical property-function relationships, ultimately enabling more effective targeting of desired functions.
Conclusion
The computational design of intrinsically disordered proteins has advanced through the integration of physics-based modeling, machine learning, and active learning strategies. These approaches enable the rational engineering of IDRs with bespoke conformational properties, binding affinities, and phase behaviors, with applications spanning biotechnology, therapeutics, and biomaterials. Continued methodological improvements and deeper understanding of sequence-function relationships will be essential for realizing the full potential of IDR design in complex biological and technological contexts.