- The paper presents a comprehensive review of GNN applications in drug discovery, detailing advances in molecular property prediction, drug-target binding, and drug repositioning.
- Key methodologies include hybrid architectures combining GNNs with techniques like XGBoost and advanced attention mechanisms to enhance predictive performance.
- The survey identifies persistent challenges such as data scarcity, model interpretability, and integration of diverse biological data, highlighting directions for future research.
Survey of Graph Neural Networks for Drug Discovery: Recent Developments and Challenges
Introduction
This survey provides a comprehensive review of the application of Graph Neural Networks (GNNs) in drug discovery, systematically categorizing recent advances into seven major research threads: molecular property prediction, drug-target binding affinity, drug-drug interaction and synergy, microbiome interaction, drug repositioning, retrosynthesis, and new drug design. The paper critically examines 38 primary research articles and four prior surveys, highlighting architectural innovations, evaluation metrics, dataset utilization, and persistent challenges in the field.
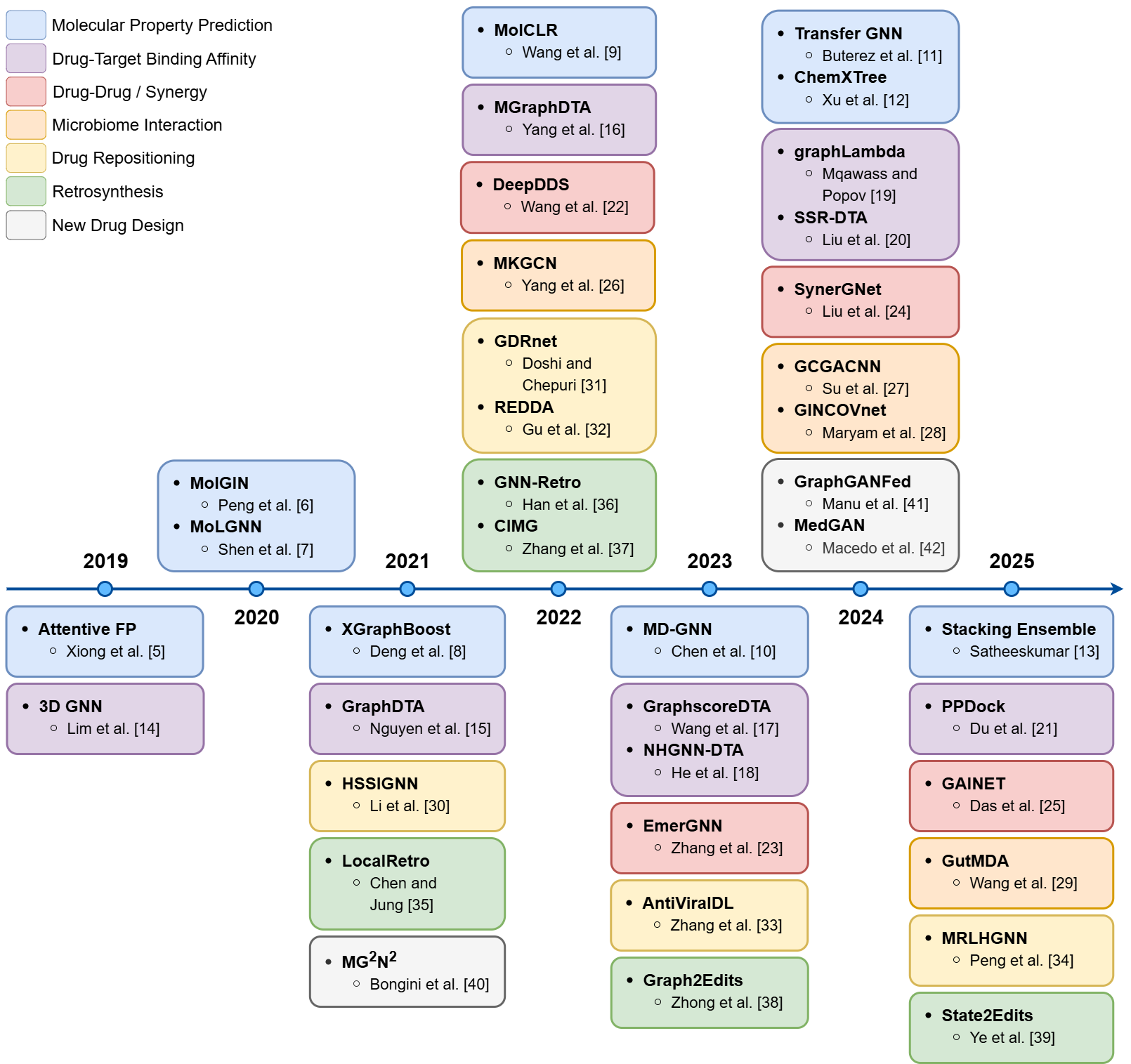
Figure 1: Timeline of major GNN-based drug discovery contributions, color-coded by research category.
GNN Architectures, Metrics, and Datasets
The survey details the evolution and deployment of core GNN architectures, including GCN, GAT, GIN, MPNN, and their variants. These models leverage message passing and attention mechanisms to encode molecular graphs, enabling the extraction of both local and global structural features. The review emphasizes the importance of architecture selection, noting that hybrid and ensemble approaches (e.g., stacking GNNs with XGBoost or integrating neural readouts) often yield superior predictive performance, especially in data-scarce regimes.
Evaluation metrics are rigorously discussed, with regression (MSE, RMSE, MAE, R2, CI), classification (AUC, F1, MCC, balanced accuracy), and generative (validity, uniqueness, novelty, QED, logP) metrics tailored to the specific drug discovery task. The survey also catalogs the most frequently used datasets, such as ESOL, FreeSolv, Lipop, BBBP, BACE, ClinTox, SIDER, Tox21, ToxCast, HIV, MUV, DUD-E, PDBbind, ChEMBL, Davis, KIBA, DrugBank, QM8, QM9, MDAD, aBiofilm, DrugVirus, Fdataset, Cdataset, KEGG, CTD, STRING, UniProt, and USPTO-50K.
Molecular Property Prediction
GNNs have demonstrated robust performance in molecular property prediction, outperforming traditional descriptor-based and deep learning models. Notable advances include Attentive FP (GAT-based, virtual supernode aggregation), MolGIN (bond feature concatenation, gated neighborhood weighting), MoLGNN (motif-level self-supervised pretraining), and hybrid models like XGraphBoost (GNN feature extraction with XGBoost). Transfer learning and contrastive learning frameworks (e.g., MolCLR, Buterez et al.) have shown strong generalization, particularly in low-fidelity or sparse data settings. Ensemble approaches (Satheeskumar) further enhance predictive accuracy and robustness, especially for ADMET/ADME tasks.
Drug-Target Binding Affinity
GNNs have become the de facto standard for drug-target binding affinity prediction, with architectures increasingly incorporating 3D structural information, hybrid protein-ligand representations, and advanced attention mechanisms. GraphDTA established a baseline by encoding drugs as graphs and proteins as sequences, while subsequent models (MGraphDTA, GraphscoreDTA, NHGNN-DTA, graphLambda, SSR-DTA, PPDock) introduced multiscale, hybrid, and equivariant GNNs, as well as interpretability modules (e.g., Grad-AAM). These models consistently outperform prior deep learning and docking methods, achieving state-of-the-art results on benchmarks such as PDBbind, Davis, KIBA, and CASF.
Drug-Drug Interaction and Synergy
GNNs have enabled accurate prediction of drug-drug interactions and synergistic effects, critical for polypharmacy and combination therapy. DeepDDS (GAT/GCN-based) and EmerGNN (flow-based, biomedical network integration) leverage heterogeneous biological data and attention mechanisms to model complex drug pair relationships. SynerGNet and GAINET further integrate protein-protein interaction networks and co-attention modules, demonstrating improved performance and interpretability over classical machine learning and knowledge graph approaches.
Microbiome Interaction
The application of GNNs to microbiome-drug interaction is emerging, with models such as MKGCN (multi-kernel fusion), GCGACNN (hybrid GCN/GAT/CNN/RF), GINCOVnet, and GutMDA (triple network integration) addressing microbe-drug association and drug susceptibility in the human microbiome. These models outperform prior methods in AUC/AUPR and demonstrate the utility of integrating biological similarity networks, though limitations in data diversity and mechanistic interpretability persist.
Drug Repositioning
GNNs have shown promise in drug repositioning by modeling heterogeneous biological networks and leveraging side information. HSSIGNN, GDRnet (SIGN-based), REDDA (multi-relation attention), AntiViralDL (LightGCN, contrastive learning), and MRLHGNN (multi-view transformer aggregation) achieve superior link prediction and drug ranking performance, facilitating the identification of novel indications and therapies. Case studies on COVID-19 and other diseases validate the practical utility of these models.
Retrosynthesis
Retrosynthesis models have evolved from template-based to semi-template and template-free GNN frameworks. LocalRetro (local/global attention), GNN-Retro (cost estimation via molecular fingerprints), CIMG (chemistry-informed graph descriptors), Graph2Edits (autoregressive graph editing), and State2Edits (state-based edit sequence prediction) demonstrate improved accuracy and scalability in reaction prediction and synthetic route planning. Integration of reaction conditions and mechanistic information remains an open challenge.
New Drug Design
Generative GNN models for new drug design are less explored but show significant potential. MG2N2 (sequential node/edge generation), GraphGANFed (GCN-GAN with federated learning), and MedGAN (WGAN-GCN for quinoline scaffolds) achieve high validity, novelty, and diversity in molecular generation. These models highlight the trade-offs between interpretability, computational efficiency, and generative quality, with federated and privacy-preserving approaches gaining traction.
Future Directions
Key challenges identified include data scarcity, model interpretability, computational complexity, and integration of heterogeneous biological information. Future work should focus on:
- Developing data-efficient and transfer learning GNNs for low-resource settings.
- Enhancing interpretability via attention visualization and motif-level explanations.
- Optimizing hybrid and ensemble architectures for scalability and accuracy.
- Expanding generative GNN frameworks for conditional and property-driven molecule design.
- Integrating microbiome and multi-omics data for holistic drug discovery.
- Addressing regulatory requirements for model transparency and reproducibility.
Conclusion
This survey synthesizes recent advances in GNN-based drug discovery, demonstrating that GNNs have become central to molecular property prediction, interaction modeling, and generative design. While state-of-the-art models achieve strong numerical results and outperform traditional approaches, persistent challenges in data, interpretability, and integration remain. Continued innovation in GNN architectures, learning paradigms, and biological data integration will be critical for realizing the full potential of AI-driven drug discovery.