Good rates from bad coordinates: the exponential average time-dependent rate approach
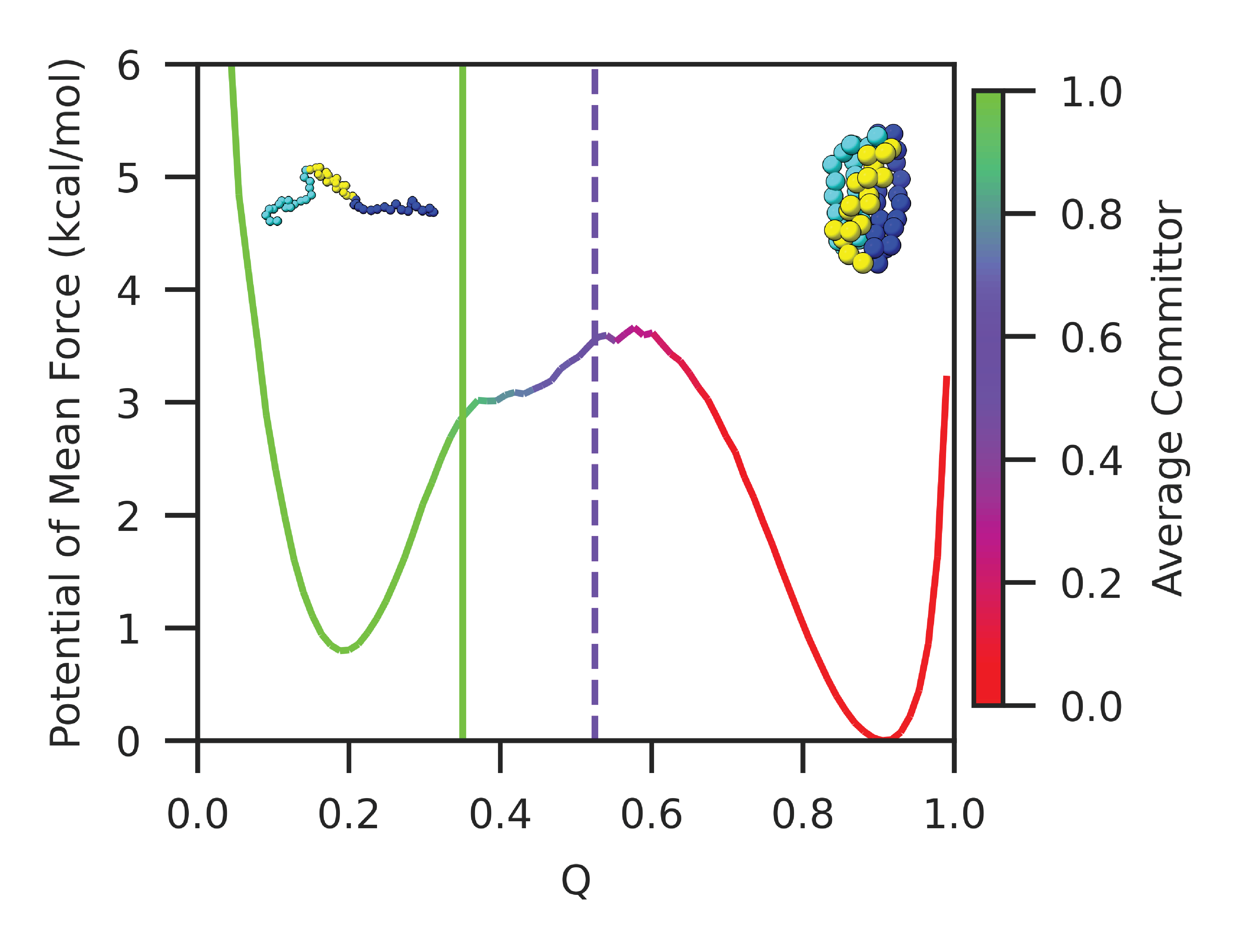
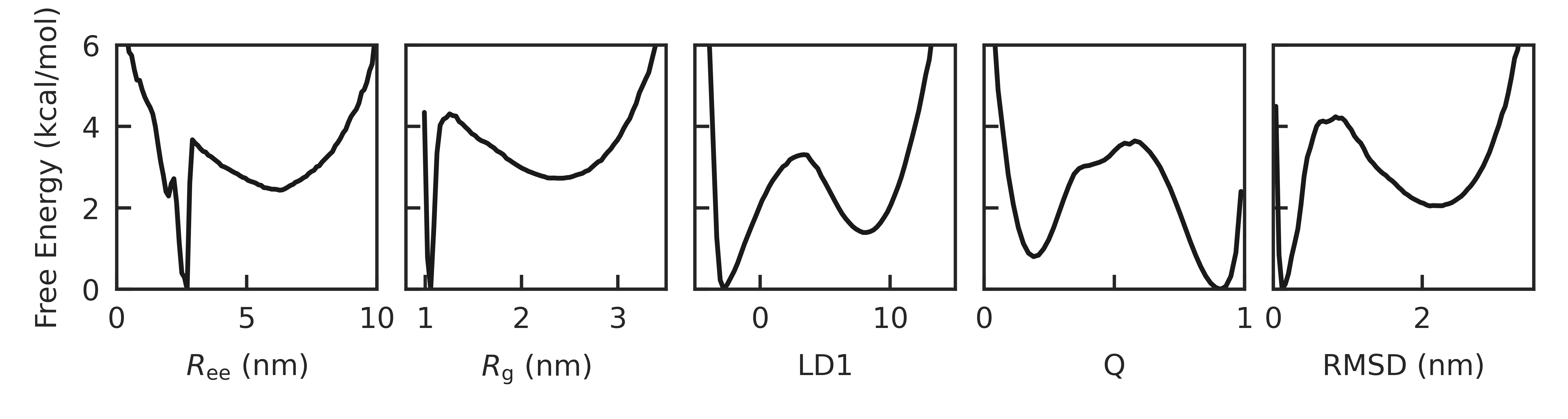
Abstract: Our ability to calculate rates of biochemical processes using molecular dynamics simulations is severely limited by the fact that the time scales for reactions, or changes in conformational state, scale exponentially with the relevant free-energy barriers. In this work, we improve upon a recently proposed rate estimator that allows us to predict transition times with molecular dynamics simulations biased to rapidly explore one or several collective variables. This approach relies on the idea that not all bias goes into promoting transitions, and along with the rate, it estimates a concomitant scale factor for the bias termed the collective variable biasing efficiency $\gamma$. First, we demonstrate mathematically that our new formulation allows us to derive the commonly used Infrequent Metadynamics (iMetaD) estimator when using a perfect collective variable, $\gamma=1$. After testing it on a model potential, we then study the unfolding behavior of a previously well characterized coarse-grained protein, which is sufficiently complex that we can choose many different collective variables to bias, but which is sufficiently simple that we are able to compute the unbiased rate dire ctly. For this system, we demonstrate that our new Exponential Average Time-Dependent Rate (EATR) estimator converges to the true rate more rapidly as a function of bias deposition time than does the previous iMetaD approach, even for bias deposition times that are short. We also show that the $\gamma$ parameter can serve as a good metric for assessing the quality of the biasing coordinate. Finally, we demonstrate that the approach works when combining multiple less-than-optimal bias coordinates.
- Chandler, D. Barrier crossings: classical theory of rare but important events. Classical and quantum dynamics in condensed phase simulations 1998, 523
- Tuckerman, M. E. Statistical mechanics: theory and molecular simulation; Oxford university press, 2023
- Thiede, E. H.; Giannakis, D.; Dinner, A. R.; Weare, J. Galerkin approximation of dynamical quantities using trajectory data. J. Chem. Phys. 2019, 150
- Guttenberg, N.; Dinner, A. R.; Weare, J. Steered transition path sampling. J. Chem. Phys. 2012, 136
- Wieczór, M.; Tang, P. K.; Orozco, M.; Cossio, P. Omicron mutations increase interdomain interactions and reduce epitope exposure in the SARS-CoV-2 spike. Iscience 2023, 26
- Peña Ccoa, W. J.; Hocky, G. M. Assessing models of force-dependent unbinding rates via infrequent metadynamics. J. Chem. Phys. 2022, 156
- Mukadum, F.; Peña Ccoa, W. J.; Hocky, G. M. Molecular simulation approaches to probing the effects of mechanical forces in the actin cytoskeleton. Cytoskeleton 2024, 1–10
- McGovern, M.; De Pablo, J. A boundary correction algorithm for metadynamics in multiple dimensions. J. Chem. Phys. 2013, 139
- Blumer, O.; Reuveni, S.; Hirshberg, B. Short-Time Infrequent Metadynamics for Improved Kinetics Inference. arXiv:2401.14237 2024,
- Kuznets-Speck, B.; Limmer, D. T. Inferring equilibrium transition rates from nonequilibrium protocols. Biophys. J. 2023,
- Cossio, P.; Hummer, G.; Szabo, A. Transition paths in single-molecule force spectroscopy. The Journal of chemical physics 2018, 148
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Collections
Sign up for free to add this paper to one or more collections.

