- The paper presents a novel MACE model that accelerates DFT phonon calculations using machine learning potentials.
- The study utilizes advanced MPNNs and a training dataset of 15,670 structures to achieve a mean absolute error of 0.18 THz in vibrational frequency predictions.
- The approach demonstrates high accuracy in predicting dynamical stability (86.2%) and thermodynamic stability across diverse material compounds.
Accelerating High-Throughput Phonon Calculations via Machine Learning Universal Potentials
Introduction
The study titled "Accelerating High-Throughput Phonon Calculations via Machine Learning Universal Potentials" presents an innovative approach to accelerating phonon calculations through machine learning-based interatomic potentials. By leveraging a Machine Learning Interatomic Potential model (MACE), the study effectively addresses the challenge of high computational costs in Density Functional Theory (DFT) phonon calculations.
Methodology and Model
The authors introduce MACE, a multi-atomic cluster expansion model, to predict the forces within crystal structures efficiently. It utilizes advanced Message-Passing Neural Networks (MPNNs) which are specialized for processing graph-structured data. The MACE model significantly reduces computational workload by predicting phonon properties with a subset of supercell structures, achieving excellent force prediction accuracy. During training, MACE employs force information alone to refine its predictions, using a training dataset of 15,670 structures derived from 2,738 crystal structures.
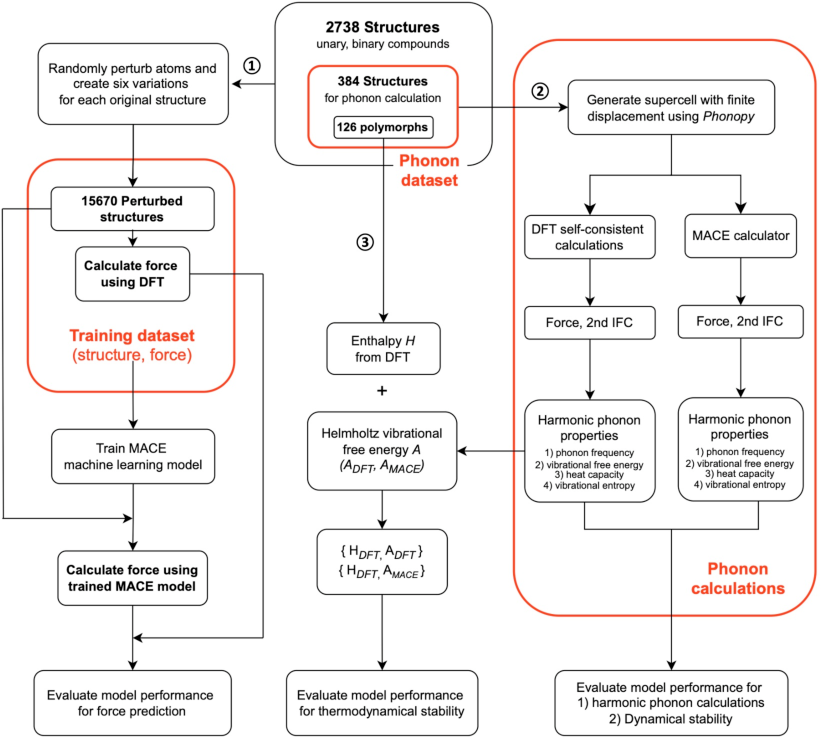
Figure 1: Workflow chart showing the computational processes employed in our study.
Dataset Construction and Training
The dataset constructed for training spans 77 elements and includes compounds of varying complexity. The data emphasizes a broad representation of forces and energies, supporting the generalizability of the model across different materials. Key to the MACE model's performance is its ability to generalize from this dataset, reducing the required number of supercell calculations typically necessary in DFT.
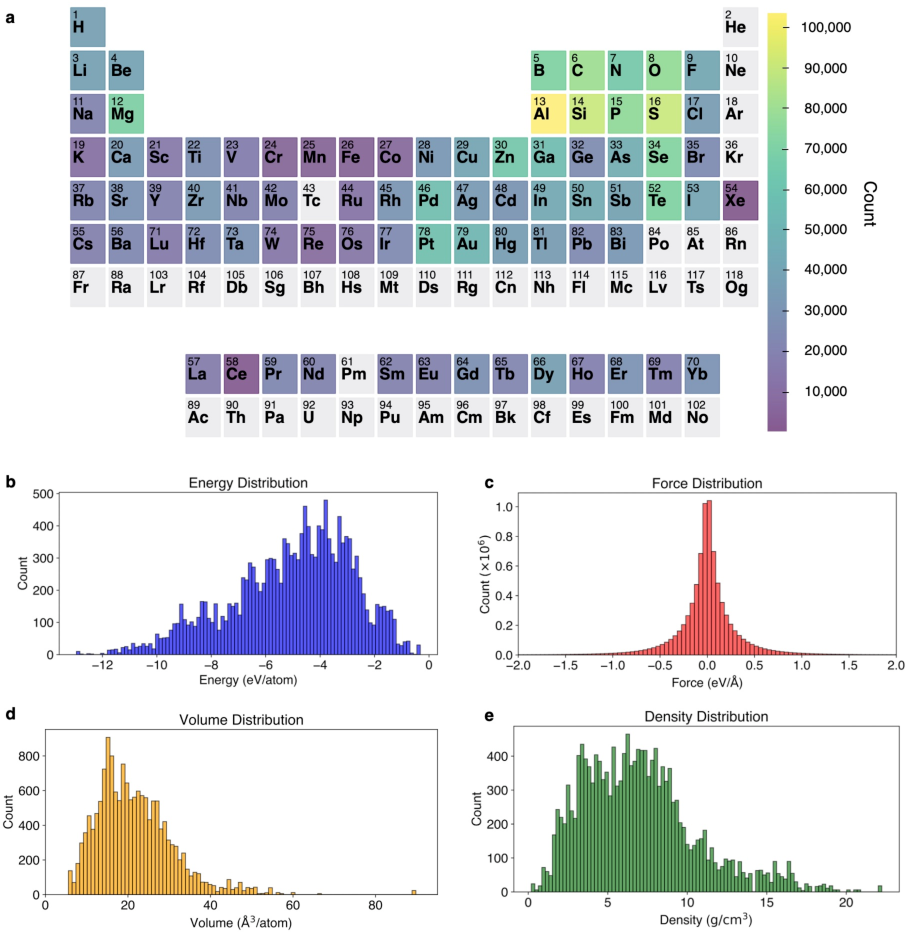
Figure 2: The heat map and distribution of elements and dataset properties crucial for model training.
Model Evaluation
The evaluation on a diverse test set of 384 materials demonstrates the MACE model's robustness in predicting phonon frequencies and thermodynamic stability. The mean absolute error (MAE) for vibrational frequencies was 0.18 THz, showcasing the MACE model’s capacity for accurate predictions despite reduced data requirements. Furthermore, the accuracy for dynamical stability predictions reached 86.2%, affirming the model's potential for identifying stable material phases.
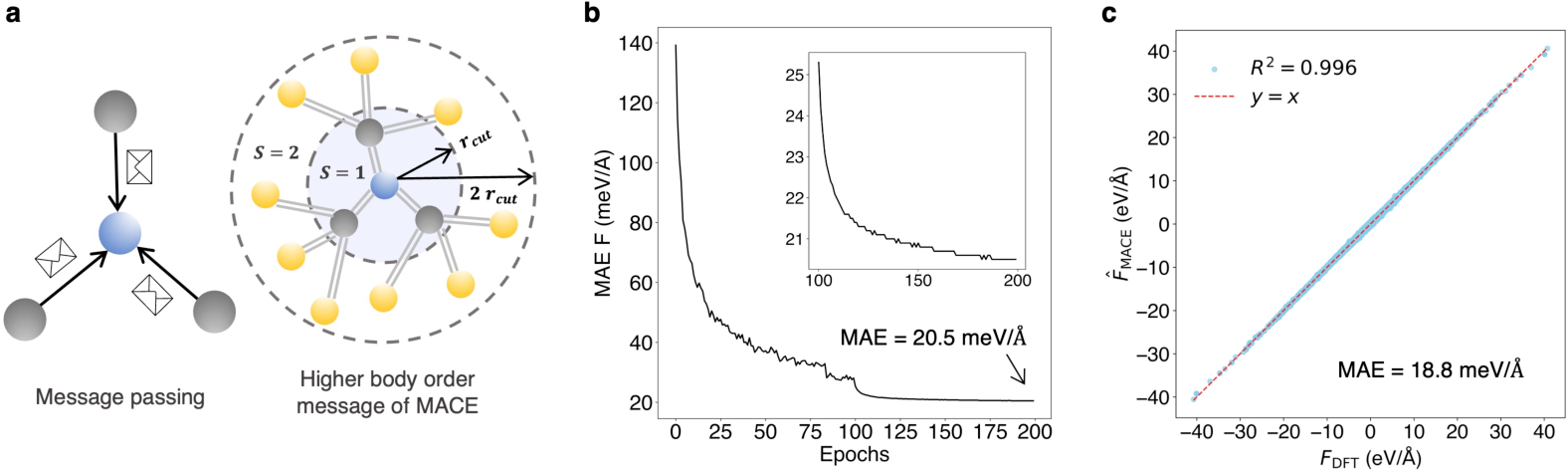
Figure 3: The MACE model's validation on force prediction, indicating strong correlation with DFT calculated forces.
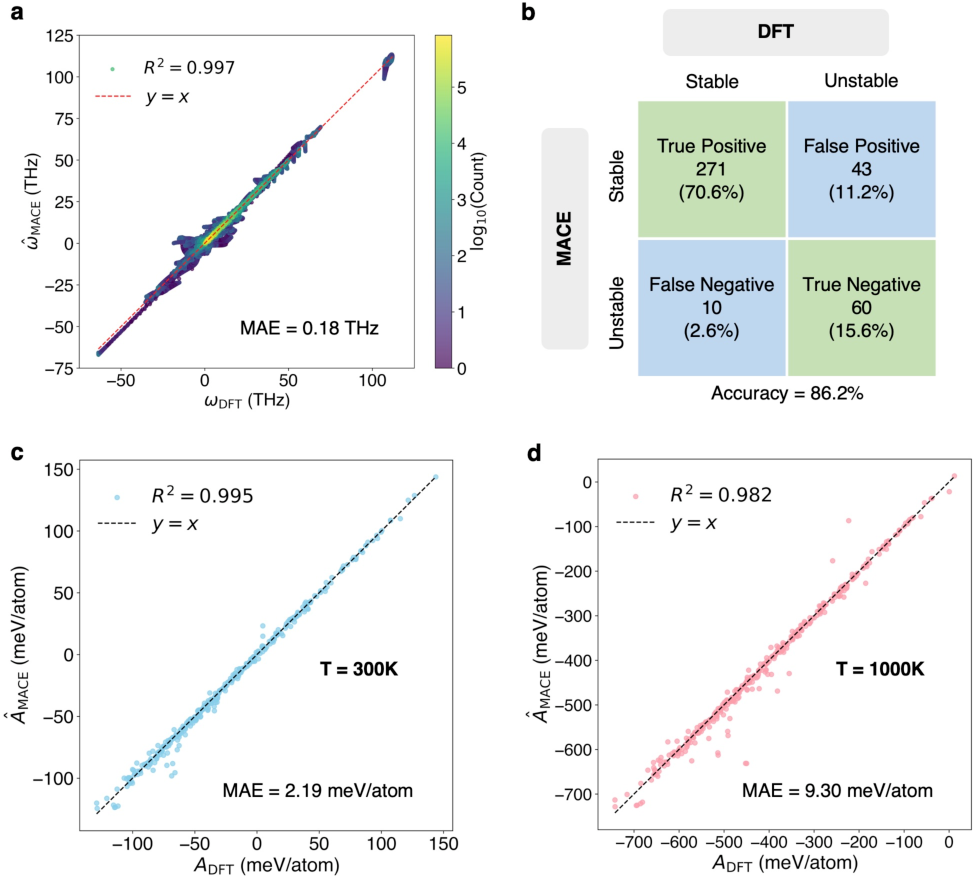
Figure 4: Evaluation of the MACE model's performance on phonon frequency prediction versus DFT calculations.
Thermodynamic Stability Analysis
The study extends the model's application to the determination of polymorphic thermodynamic stability, leveraging Helmholtz free energy calculations. The analysis covers 126 polymorphs across 49 distinct types, revealing consistent predictions from both MACE and DFT methods. This comparative study has implications for predicting phase stability under varying thermodynamic conditions.
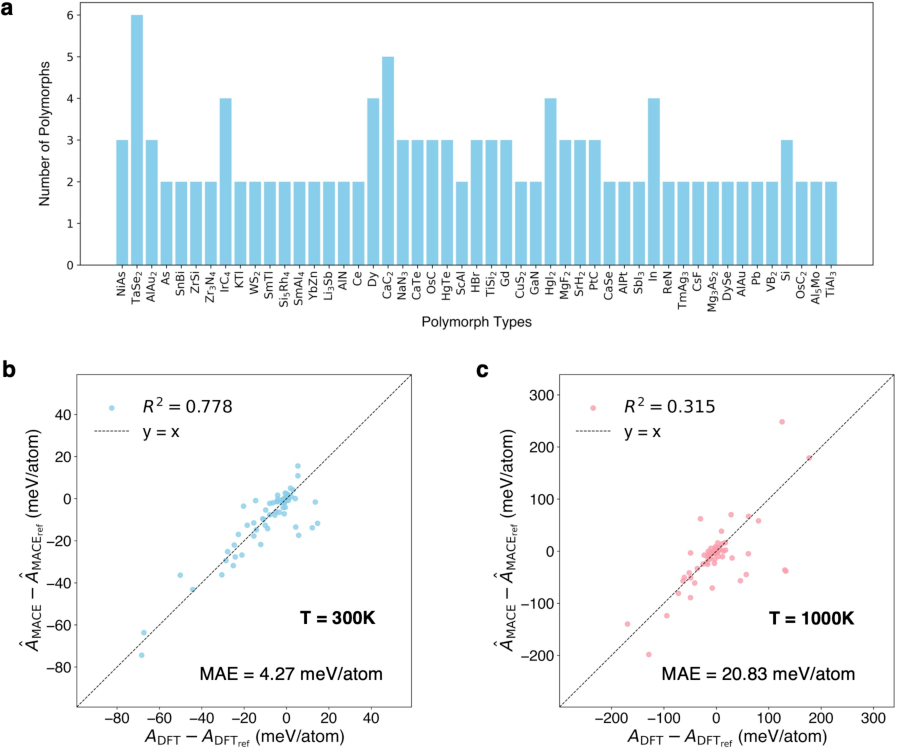
Figure 5: Thermodynamic stability of polymorphs within the phonon dataset.
Conclusion
This research highlights the efficacy of machine learning in accelerating computationally expensive DFT calculations for phonon properties. The MACE model, trained on a systematically constructed dataset, proves to be a powerful alternative, mitigating the computational demand traditionally required by DFT in high-throughput materials screening. Future work may include expanding the methodology to incorporate anharmonic effects and extending the dataset to include more complex compounds. The insights garnered have broad implications, potentially impacting fields of material discovery and computational materials science at large.