- The paper presents a novel stationary-action framework that minimizes time non-local action to optimize memory kernels and noise terms in CG models.
- It integrates the Mori-Zwanzig formalism with iterative Boltzmann inversion to ensure dynamic properties like VACF and diffusion closely match all-atom references.
- The practical application to water molecules using the SPC/E model demonstrates significantly improved dynamical fidelity in coarse-grained simulations.
Summary of "Coarse-grained Mori-Zwanzig dynamics in a time-non-local stationary-action framework"
This paper presents a novel approach to address the common issue of loss in dynamical consistency when implementing coarse-grained (CG) models in computational simulations. The focus is on enhancing the ability of CG models to reproduce the time scales and dynamical properties of their corresponding all-atom reference systems. The method is based on a time-non-local stationary-action framework informed by the Mori-Zwanzig (MZ) formalism, leading to the parametrization of generalized Langevin equations (GLEs) for CG systems.
Theoretical Framework and Methodology
The MZ formalism is a cornerstone in understanding the mapping of detailed atomistic models to their coarse-grained counterparts. The CG model inherits a set of generalized Langevin equations which include memory and noise terms, representing the influence of fast atomistic dynamics integrated out during the coarse-graining process. This study proposes using a stationary-action principle, valid across both all-atom and CG representations, as a means to parameterize these terms. The minimization of a time non-local action functional is used to optimize the GLE parameters, particularly the memory kernel and noise, making this approach computationally viable.
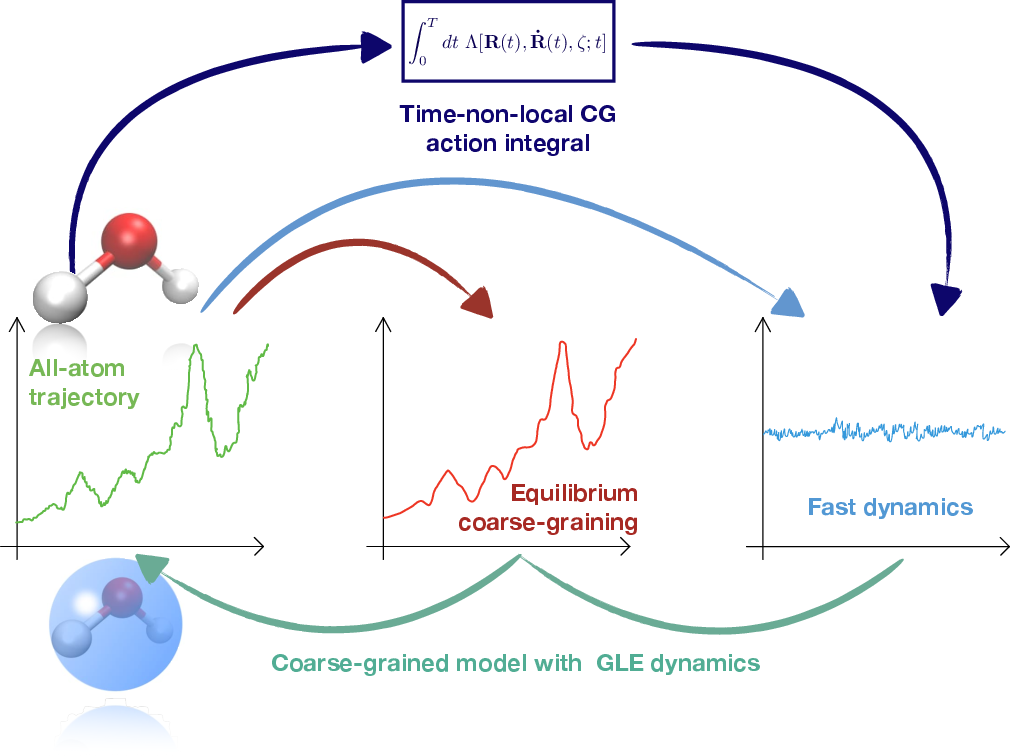
Figure 1: The proposed method integrates fast dynamics into CG models through optimized memory and noise terms recovered via a stationary-action framework.
Application to Water Molecules
A practical application of the method is demonstrated using a system of water molecules. The water molecules, represented by a three-point SPC/E model, were subjected to microcanonical ensemble simulations. These results provide a baseline for constructing an optimized CG model where each water molecule is reduced to its center of mass. The iterative Boltzmann inversion technique was employed to derive the effective CG potential from the atomistic radial distribution function. Subsequently, the GLE parameters were optimized using the action minimization approach, showcasing improved dynamical fidelity in CG simulations.
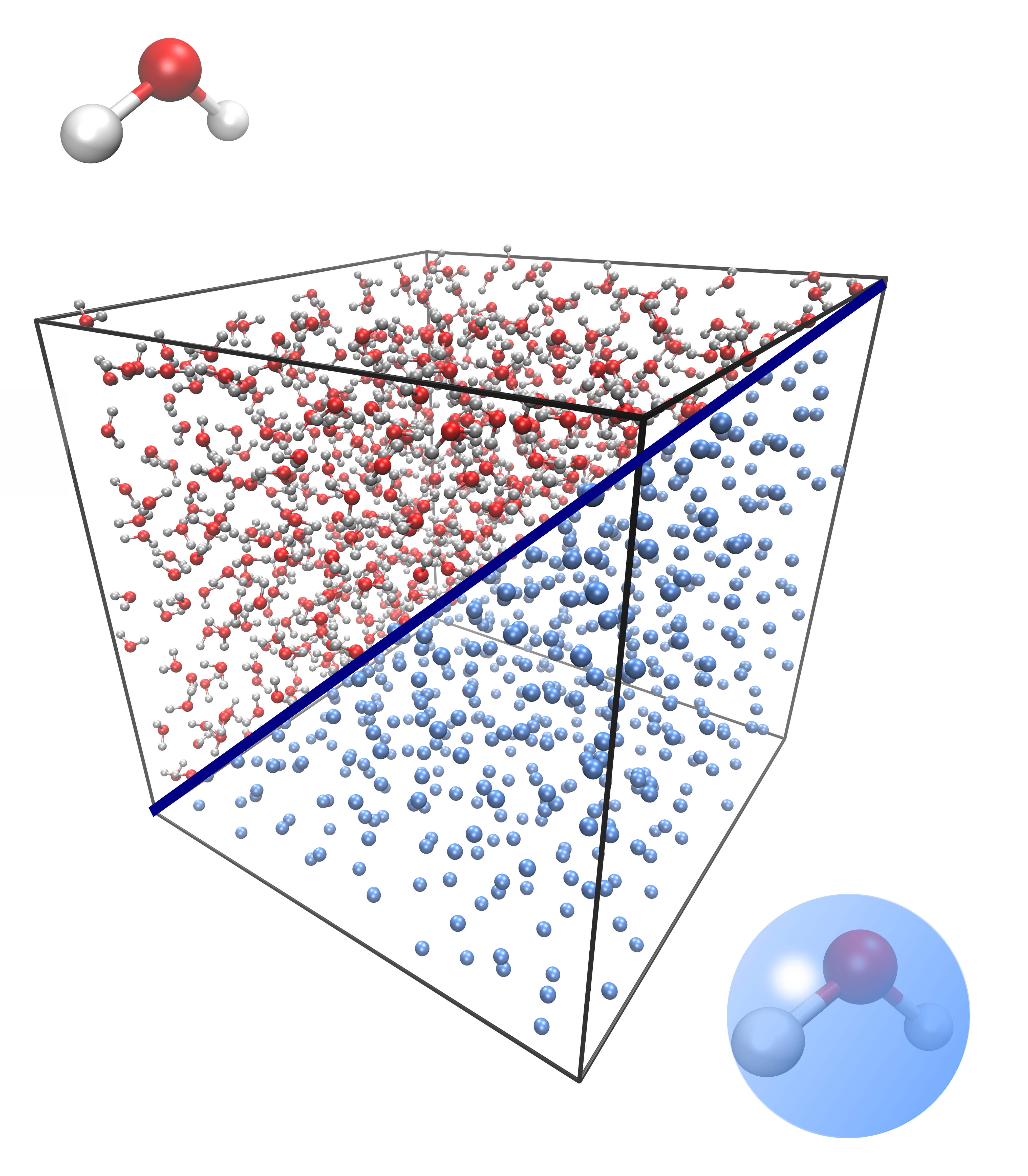
Figure 2: Atomistic and coarse-grained representations of a water system, illustrating the single CG bead mapping per water molecule.
Results and Discussion
Key metrics such as the velocity autocorrelation function (VACF) and diffusion coefficients reinforce the effectiveness of the proposed method. Notably, the VACF obtained from the GLE simulation closely matched the all-atom reference, outperforming traditional Langevin-based CG simulations. This suggests that the integration of optimized memory and noise terms leads to better retention of dynamical features.
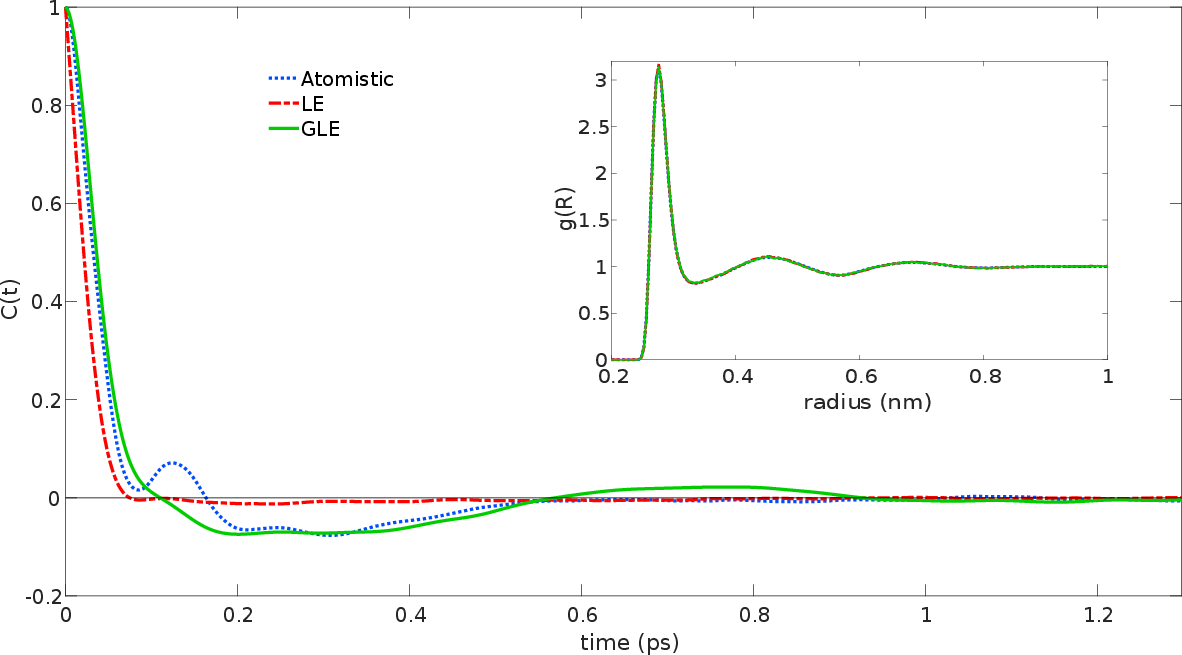
Figure 3: VACF and RDF comparisons between atomistic and CG simulated data, demonstrating the effectiveness of GLE in capturing accurate dynamic properties.
Conclusions and Implications
The stationary-action framework developed in this study provides a robust mechanism to enhance the consistency of dynamical properties in CG models, bridging the gap to their all-atom origins. This methodology not only maintains structural accuracy but also dramatically improves dynamical matching, paving the way for more reliable CG representations in complex systems. Future work may involve generalized applications across varied molecular systems and consideration of multinomial models within this theoretical context.
The implications of this research extend to the design and parametrization of CG models in diverse fields, potentially enhancing simulation accuracy for systems requiring long-time dynamic predictions. Moreover, this approach may inform new strategies in the development and analysis of complex soft-matter systems, fostering advancements in materials science, biophysics, and related domains.