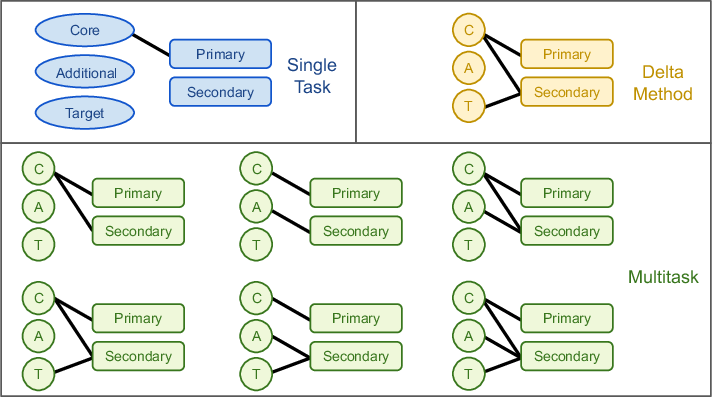
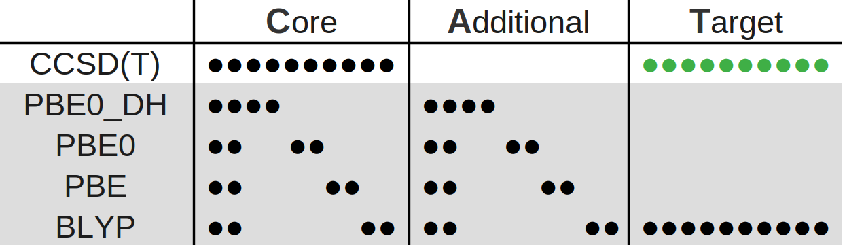
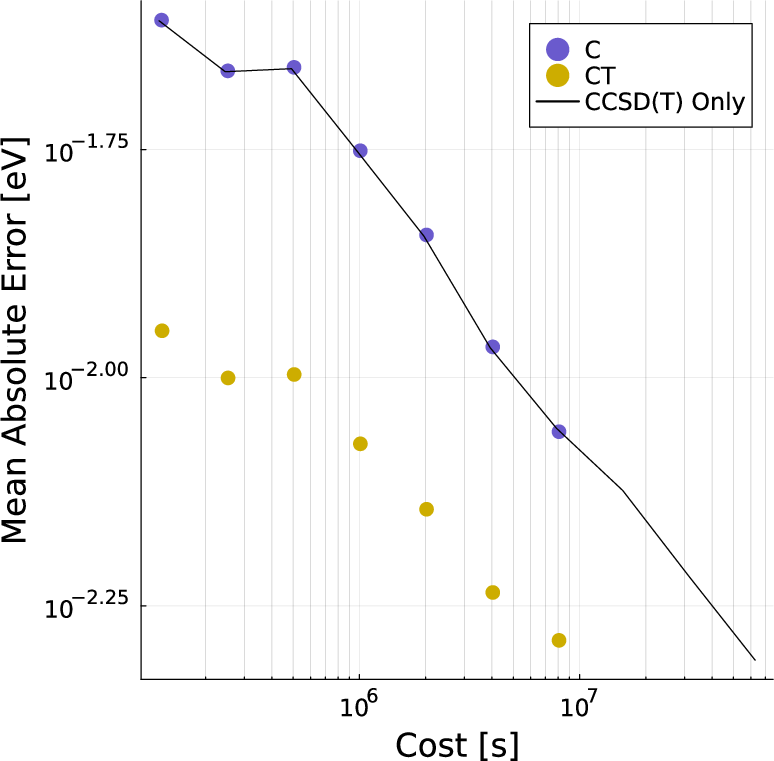
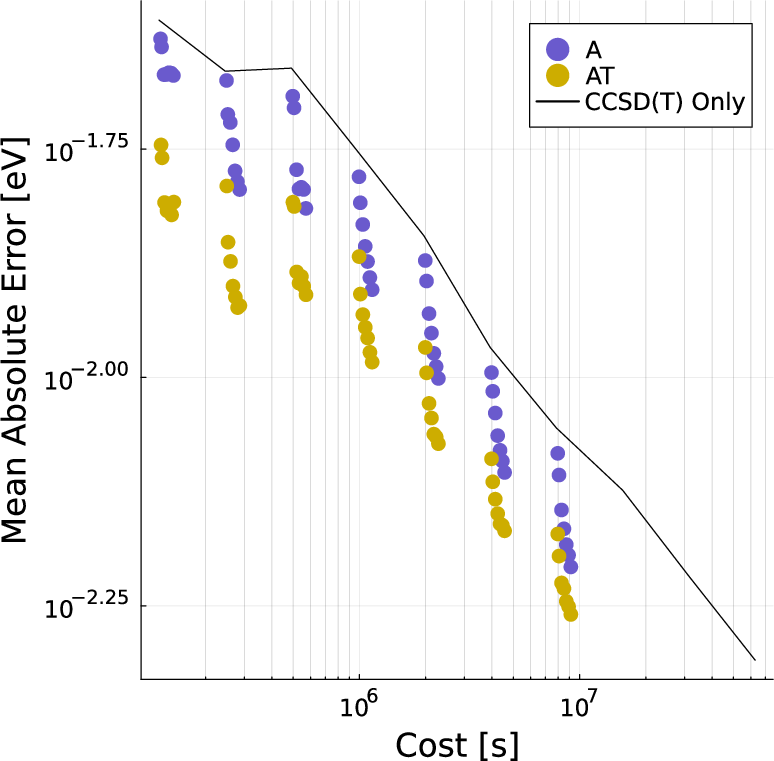
Multitask methods for predicting molecular properties from heterogeneous data (2401.17898v2)
Abstract: Data generation remains a bottleneck in training surrogate models to predict molecular properties. We demonstrate that multitask Gaussian process regression overcomes this limitation by leveraging both expensive and cheap data sources. In particular, we consider training sets constructed from coupled-cluster (CC) and density functional theory (DFT) data. We report that multitask surrogates can predict at CC-level accuracy with a reduction to data generation cost by over an order of magnitude. Of note, our approach allows the training set to include DFT data generated by a heterogeneous mix of exchange-correlation functionals without imposing any artificial hierarchy on functional accuracy. More generally, the multitask framework can accommodate a wider range of training set structures -- including full disparity between the different levels of fidelity -- than existing kernel approaches based on $\Delta$-learning, though we show that the accuracy of the two approaches can be similar. Consequently, multitask regression can be a tool for reducing data generation costs even further by opportunistically exploiting existing data sources.
- M. E. Harding, T. Metzroth, J. Gauss, and A. A. Auer, “Parallel calculation of ccsd and ccsd(t) analytic first and second derivatives,” Journal of Chemical Theory and Computation 4, 64–74 (2008).
- L. Ying, J. Yu, and L. Ying, “Numerical methods for kohn–sham density functional theory,” Acta Numerica 28, 405–539 (2019).
- J. Perdew and K. Schmidt, “Jacob’s ladder of density functional approximations for the exchange-correlation energy,” in AIP Conference Proceedings (2001) presented at AIP Conference Proceedings 577.
- L. Goerigkab and S. Grimme, “A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions,” Physical Chemistry Chemical Physics 13, 6670–6688 (2011).
- A. M. Teale, T. Helgaker, A. Savin, C. Adano, B. Aradi, A. V. Arbuznikov, P. Ayers, E. J. Baerends, V. Barone, P. Calaminici, E. Cances, E. A. Carter, P. K. Chattaraj, H. Chermette, I. Ciofini, T. D. Crawford, F. De Proft, J. Dobson, C. Draxl, T. Frauenheim, E. Fromager, P. Fuentealba, L. Gagliardi, G. Galli, J. Gao, P. Geerlings, N. Gidopoulos, P. M. W. Gill, P. Gori-Giorgi, A. Görling, T. Gould, S. Grimme, O. Gritsenko, H. J. A. Jensen, E. R. Johnson, R. O. Jones, M. Kaupp, A. Koster, L. Kronik, A. I. Krylov, S. Kvaal, A. Laestadius, M. P. Levy, M. Lewin, S. Liu, P.-F. Loos, N. T. Maitra, F. Neese, J. Perdew, K. Pernal, P. Pernot, P. Piecuch, E. Rebolini, L. Reining, P. Romaniello, A. Ruzsinszky, D. Salahub, M. Scheffler, P. Schwerdtfeger, V. N. Staroverov, J. Sun, E. Tellgren, D. J. Tozer, S. Trickey, C. A. Ullrich, A. Vela, G. Vignale, T. A. Wesolowski, X. Xu, and W. Yang, “Dft exchange: Sharing perspectives on the workhorse of quantum chemistry and materials science,” Physical Chemistry Chemical Physics (2022), 10.1039/d2cp02827a.
- J. S. Smith, B. T. Nebgen, R. Zubatyuk, N. Lubbers, C. Devereux, K. Barros, S. Tretiak, O. Isayev, and A. E. Roitberg, “Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning,” Nature Communications 10 (2019), 10.1038/s41467-019-10827-4.
- P. O. Dral, A. Owens, A. Dral, and G. Csányi, “Hierarchical machine learning of potential energy surfaces,” The Journal of Chemical Physics 152 (2020), 10.1063/5.0006498.
- S. M. Goodlett, J. M. Turney, and H. F. Schaefer, “Comparison of multifidelity machine learning models for potential energy surfaces,” The Journal of Chemical Physics 159 (2023), 10.1063/5.0158919.
- V. Zaverkin, D. Holzmüller, L. Bonfirraro, and J. Kästner, “Transfer learning for chemically accurate interatomic neural network potentials,” Physical Chemistry Chemical Physics 25, 5383–5396 (2023).
- S. Curtarolo, W. Setyawan, G. L. Hart, M. Jahnatek, R. V. Chepulskii, R. H. Taylor, S. Wang, J. Xue, K. Yang, O. Levy, M. J. Mehl, H. T. Stokes, D. O. Demchenko, and D. Morgan, “Aflow: An automatic framework for high-throughput materials discovery,” Computational Materials Science 58, 218–226 (2012).
- A. Jain, G. Hautier, C. J. Moore, S. Ping Ong, C. C. Fischer, T. Mueller, K. A. Persson, and G. Ceder, “A high-throughput infrastructure for density functional theory calculations,” Computational Materials Science 50, 2295–2310 (2011).
- S. P. Huber, S. Zoupanos, M. Uhrin, L. Talirz, L. Kahle, R. Häuselmann, D. Gresch, T. Müller, A. V. Yakutovich, C. W. Andersen, F. F. Ramirez, C. S. Adorf, F. Gargiulo, S. Kumbhar, E. Passaro, C. Johnston, A. Merkys, A. Cepellotti, N. Mounet, N. Marzari, B. Kozinsky, and G. Pizzi, “Aiida 1.0, a scalable computational infrastructure for automated reproducible workflows and data provenance,” Scientific Data 7 (2020), 10.1038/s41597-020-00638-4.
- E. Cancès, A. Levitt, Y. Maday, and C. Yang, “Numerical methods for kohn–sham models: Discretization, algorithms, and error analysis,” in Density Functional Theory: Modeling, Mathematical Analysis, Computational Methods, and Applications (Springer, 2022) pp. 333–400.
- M. F. Herbst and A. Levitt, “Black-box inhomogeneous preconditioning for self-consistent field iterations in density functional theory,” Journal of Physics: Condensed Matter (2020), 10.1088/1361-648x/abcbdb.
- M. F. Herbst and A. Levitt, “A robust and efficient line search for self-consistent field iterations,” Journal of Computational Physics 459, 111127 (2022).
- E. Cancès, M. F. Herbst, G. Kemlin, A. Levitt, and B. Stamm, “Numerical stability and efficiency of response property calculations in density functional theory,” Letters in Mathematical Physics 113 (2023), 10.1007/s11005-023-01645-3.
- L. Ruddigkeit, R. van Deursen, L. C. Blum, and J.-L. Reymond, “Enumeration of 166 billion organic small molecules in the chemical universe database gdb-17,” Journal of Chemical Information and Modeling 52, 2864–2875 (2012), pMID: 23088335, https://doi.org/10.1021/ci300415d .
- R. Ramakrishnan, P. O. Dral, M. Rupp, and O. A. von Lilienfeld, “Quantum chemistry structures and properties of 134 kilo molecules,” Scientific Data 1 (2014).
- L. Chanussot*, A. Das*, S. Goyal*, T. Lavril*, M. Shuaibi*, M. Riviere, K. Tran, J. Heras-Domingo, C. Ho, W. Hu, A. Palizhati, A. Sriram, B. Wood, J. Yoon, D. Parikh, C. L. Zitnick, and Z. Ulissi, “Open catalyst 2020 (oc20) dataset and community challenges,” ACS Catalysis (2021), 10.1021/acscatal.0c04525.
- J. Smith, O. Isayev, and A. Roitberg, “A data set of 20 million calculated off-equilibrium conformations for organic molecules,” Scientific Data 4 (2017).
- E. Bonilla, K. Chai, and C. Williams, “Multi-task gaussian process prediction,” in Advances in neural information processing systems, edited by J. Platt, D. Koller, Y. Singer, and S. Roweis (MIT Press, Cambridge, Massachusetts, 2008) pp. 153–160.
- G. Leen, J. Peltonen, and S. Kaski, “Focused multi-task learning in a gaussian process framework,” Machine Learning 1-2, 157–182 (2012).
- G. Pilania, J. Gubernatis, and T. Lookman, “Multi-fidelity machine learning models for accurate bandgap predictions of solids,” Computational Materials Science 129, 156–162 (2017).
- R. Batra, G. Pilania, B. Uberuaga, and R. Ramprasad, “Multifidelity information fusion with machine learning: A case study of dopant formation energies in hafnia,” ACS Applied Materials & Inference 11, 24906–24918 (2019).
- M. Kennedy and A. O’Hagan, “Predicting the output from a complex computer code when fast approximations are available,” Biometrika 87, 1–13 (2000).
- R. Ramakrishnan, P. O. Dral, M. Rupp, and O. A. von Lilienfeld, “Big data meets quantum chemistry approximations: The ΔΔ\Deltaroman_Δ-machine learning approach,” Journal of Chemical Theory and Computation 11, 2087–2096 (2015).
- P. O. Dral, T. Zubatiuk, and B.-X. Xue, “Learning from multiple quantum chemical methods: δ𝛿\deltaitalic_δ-learning, transfer learning, co-kriging, and beyond,” in Quantum Chemistry in the Age of Machine Learning (Elsevier, 2023) pp. 491–507.
- V. Vinod, U. Kleinekathöfer, and P. Zaspel, “Optimized multifidelity machine learning for quantum chemistry,” (2023), arXiv:2312.05661 [physics.chem-ph] .
- G. G. Towell and J. W. Shavlik, “Knowledge-based artificial neural networks,” Artificial Intelligence 70, 119–165 (1994).
- L.-M. Fu, “Integration of neural heuristics into knowledge-based inference,” in International 1989 Joint Conference on Neural Networks (1989) pp. 606 vol.2–.
- P. L. Bartlett, A. Montanari, and A. Rakhlin, “Deep learning: a statistical viewpoint,” Acta Numerica 30, 87–201 (2021).
- S. Lotfi, M. Finzi, S. Kapoor, A. Potapczynski, M. Goldblum, and A. G. Wilson, “Pac-bayes compression bounds so tight that they can explain generalization,” (2022), arXiv:2211.13609 [cs.LG] .
- C. E. Rasmussen and C. K. I. Williams, Gaussian Processes for Machine Learning (the MIT Press, 2006).
- J. Quinonero-Candela and C. Rasmussen, “A unifying view of sparse approximate gaussian process regression,” Journal of Machine Learning Research 6, 1939–1959 (2005).
- H. Liu, Y.-S. Ong, X. Shen, and J. Cai, “When gaussian process meets big data: A review of scalable gps,” (2018).
- A. G. Wilson, C. Dann, and H. Nickisch, “Thoughts on massively scalable gaussian processes,” CoRR abs/1511.01870 (2015), 1511.01870 .
- D. A. Cole, R. B. Christianson, and R. B. Gramacy, “Locally induced gaussian processes for large-scale simulation experiments,” Statistics and Computing 31, 1573–1375 (2021).
- P. Abrahamsen, “A review of gaussian random fields and correlation functions,” (1997), 10.13140/RG.2.2.23937.20325.
- A. P. Bartók, M. C. Payne, R. Kondor, and G. Csányi, “Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons,” Physical Review Letters 104 (2010), 10.1103/physrevlett.104.136403.
- A. P. Bartók, R. Kondor, and G. Csányi, “On representing chemical environments,” Physical Review B 87, 184115 (2013).
- A. P. Bartók and G. Csányi, “Gaussian approximation potentials: A brief tutorial introduction,” International Journal of Quantum Chemistry 115, 1051–1057 (2015).
- M. Chen, H.-Y. Ko, R. Remsing, M. Andrade, B. Santra, Z. Sun, A. Selloni, R. Car, M. Klein, J. Perdew, and W. Xifan, “Ab initio theory and modeling of water,” Proceedings of the National Academy of Sciences 114, 201712499 (2017).
- S. Dasgupta, E. Lambros, J. Perdew, and F. Paesani, “Elevating density functional theory to chemical accuracy for water simulations through a density-corrected many-body formalism,” (2021), 10.33774/chemrxiv-2021-hstgf.
- M. J. Gillan, D. Alfè, and A. Michaelides, “Perspective: How good is DFT for water?” The Journal of Chemical Physics 144, 130901 (2016), https://pubs.aip.org/aip/jcp/article-pdf/doi/10.1063/1.4944633/13696839/130901_1_online.pdf .
- Q. Yu, C. Qu, P. L. Houston, R. Conte, A. Nandi, and J. M. Bowman, “q-aqua: A many-body ccsd(t) water potential, including four-body interactions, demonstrates the quantum nature of water from clusters to the liquid phase,” The Journal of Physical Chemistry Letters 13, 5068–5074 (2022), pMID: 35652912.
- D. G. A. Smith, L. A. Burns, A. C. Simmonett, R. M. Parrish, M. C. Schieber, R. Galvelis, P. Kraus, H. Kruse, R. Di Remigio, A. Alenaizan, A. M. James, S. Lehtola, J. P. Misiewicz, M. Scheurer, R. A. Shaw, J. B. Schriber, Y. Xie, Z. L. Glick, D. A. Sirianni, J. S. O’Brien, J. M. Waldrop, A. Kumar, E. G. Hohenstein, B. P. Pritchard, B. R. Brooks, I. Schaefer, Henry F., A. Y. Sokolov, K. Patkowski, I. DePrince, A. Eugene, U. Bozkaya, R. A. King, F. A. Evangelista, J. M. Turney, T. D. Crawford, and C. D. Sherrill, “PSI4 1.4: Open-source software for high-throughput quantum chemistry,” The Journal of Chemical Physics 152, 184108 (2020), https://pubs.aip.org/aip/jcp/article-pdf/doi/10.1063/5.0006002/16684807/184108_1_online.pdf .
- J. P. Perdew, K. Burke, and M. Ernzerhof, “Generalized gradient approximation made simple,” Phys. Rev. Lett. 77, 3865–3868 (1996).
- J. Sun, A. Ruzsinszky, and J. P. Perdew, “Strongly constrained and appropriately normed semilocal density functional,” Phys. Rev. Lett. 115, 036402 (2015).
- A. J. A. Price, K. R. Bryenton, and E. R. Johnson, “Requirements for an accurate dispersion-corrected density functional,” The Journal of Chemical Physics 154, 230902 (2021).
- X. Huang, B. J. Braams, and J. M. Bowman, “Ab initio potential energy and dipole moment surfaces of (h2o)2,” The Journal of Physical Chemistry A 110, 445–451 (2006), pMID: 16405316, https://doi.org/10.1021/jp053583d .
- C. Duan, K. Fisher, and H. Kulik, “kefisher98/IP_EA_deltaSCF: Ionization Potential, Electron Affinity, and Delta SCF for Small Organic Molecules,” (2023).
- C. Duan, F. Liu, A. Nandy, and H. J. Kulik, “Data-driven approaches can overcome the cost–accuracy trade-off in multireference diagnostics,” Journal of Chemical Theory and Computation 16, 4373–4387 (2020).
- C. Duan, S. Chen, M. G. Taylor, F. Liu, and H. J. Kulik, “Machine learning to tame divergent density functional approximations: a new path to consensus materials design principles,” Chem. Sci. 12, 13021–13036 (2021).
- E. Brémond and C. Adamo, “Seeking for parameter-free double-hybrid functionals: The PBE0-DH model,” The Journal of Chemical Physics 135, 024106 (2011).
- C. Adamo and V. Barone, “Toward reliable density functional methods without adjustable parameters: The PBE0 model,” J. Chem. Phys. 110, 6158–6170 (1999).
- C. Lee, W. Yang, and R. G. Parr, “Development of the colle-salvetti correlation-energy formula into a functional of the electron density,” Phys. Rev. B 37, 785–789 (1988).
- S. De, A. P. Bartók, G. Csányi, and M. Ceriotti, “Comparing molecules and solids across structural and alchemical space,” Physical Chemistry Chemical Physics 18, 13754–13769 (2016).
- V. Deringer, A. Bartók, N. Bernstein, D. Wilkins, M. Ceriotti, and G. Csányi, “Gaussian process regression for materials and modelling,” Chemical Reviews 121, 10073–10041 (2021).
- F. Musil, A. Grisafi, A. P. Bartók, C. Ortner, G. Csányi, and M. Ceriotti, “Physics-inspired structural representations for molecules and materials,” Chemical Reviews 121, 9759–9815 (2021).
- A. I. J. Forrester and A. J. K. A. Sóbester, “Multi-fidelity optimization via surrogate modelling,” Proceedings of Royal Society A 463, 3251–3269 (2007).
- A. Reuther, J. Kepner, C. Byun, S. Samsi, W. Arcand, D. Bestor, B. Bergeron, V. Gadepally, M. Houle, M. Hubbell, M. Jones, A. Klein, L. Milechin, J. Mullen, A. Prout, A. Rosa, C. Yee, and P. Michaleas, “Interactive supercomputing on 40,000 cores for machine learning and data analysis,” in 2018 IEEE High Performance extreme Computing Conference (HPEC) (IEEE, 2018) pp. 1–6.
- B. Civalleri, D. Presti, R. Dovesi, and A. Savin, “On choosing the best density functional approximation,” in Uncertainty Quantification in Multiscale Materials Modeling, edited by M. Springborg (RSC Publishing, 2012) Chap. 6, pp. 168–185.
- L. Himanen, M. O. J. Jäger, E. V. Morooka, F. Federici Canova, Y. S. Ranawat, D. Z. Gao, P. Rinke, and A. S. Foster, “DScribe: Library of descriptors for machine learning in materials science,” Computer Physics Communications 247, 106949 (2020).