- The paper introduces PPDiff, a diffusion model that integrates hybrid sequence-structure optimization for enhanced protein complex design.
- It employs a Sequence Structure Interleaving Network with kNN graph layers and causal attention to capture both global and local amino acid interactions.
- PPDiff achieved up to 50% success rates (ipTM > 0.8) and outperformed baseline methods in mini-binder and antigen-antibody complex designs.
"PPDiff: Diffusing in Hybrid Sequence-Structure Space for Protein-Protein Complex Design" (2506.11420)
Introduction
The research paper introduces PPDiff, a model designed to tackle the intricate problem of designing high-affinity protein-binding proteins for arbitrary targets. The difficulty of designing such proteins is underscored by the limitations observed in traditional empirical methods and emerging deep learning techniques. These approaches often require extensive wet-lab resources and struggle with low success rates due to sequence-structure mismatches and limited adaptability to diverse protein targets. PPDiff incorporates a novel approach by employing a diffusion model combined with a Sequence Structure Interleaving Network (SSINC) to enhance the design of protein-protein interactions.
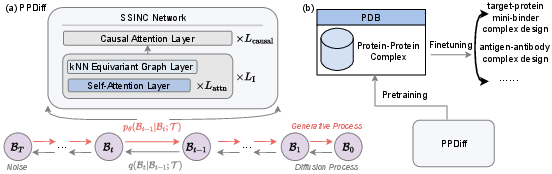
Figure 1: (a) Overall architecture of our proposed PPDiff. (b) Pretraining and application framework for protein-protein complex design.
Methodology
PPDiff leverages the strengths of diffusion models in generating protein complexes by performing simultaneous sequence and structure optimization in a non-autoregressive fashion. The model uses SSINC, which integrates interleaved self-attention layers with k-nearest neighbor (kNN) equivariant graph layers to capture both global and local amino acid interactions. A causal attention layer further simplifies the interdependencies within protein sequences, allowing for efficient noise adjustment in the diffusion process.
The training process involves pretraining on a curated dataset, PPBench, consisting of 706,360 protein complexes, followed by finetuning the model on specific real-world design tasks such as target-protein mini-binder and antigen-antibody complex designs. The efficacy of PPDiff is evaluated using metrics such as ipTM, pTM, PAE, and pLDDT, with the performance indicating notable improvements over existing methods.
Results
General Protein-Protein Complex Design
In the general design task, PPDiff achieved success rates and statistical measures that outperformed foundational models. Top candidate complexes consistently met high standards, with success rates reaching 50.00\% for ipTM scores above 0.8, highlighting the model's ability to navigate the sequence-structure landscape effectively.
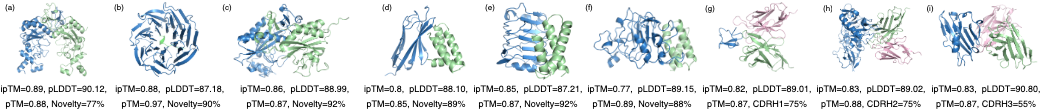
Figure 2: Designed protein complexes showing high-affinity binding across diverse scaffolds.
Real-World Applications
In target-protein mini-binder design, PPDiff demonstrated a success rate of 23.16\%, significantly higher than baseline techniques. Similarly, for the antigen-antibody complex design task, the model maintained a strong performance, reinforcing its applicability in designing novel and effective binders across varied interfaces.
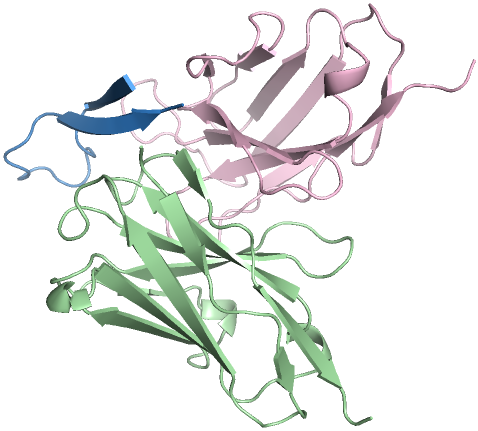
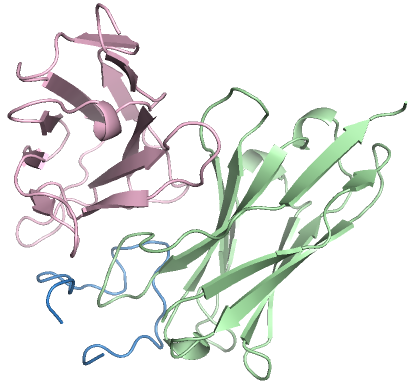
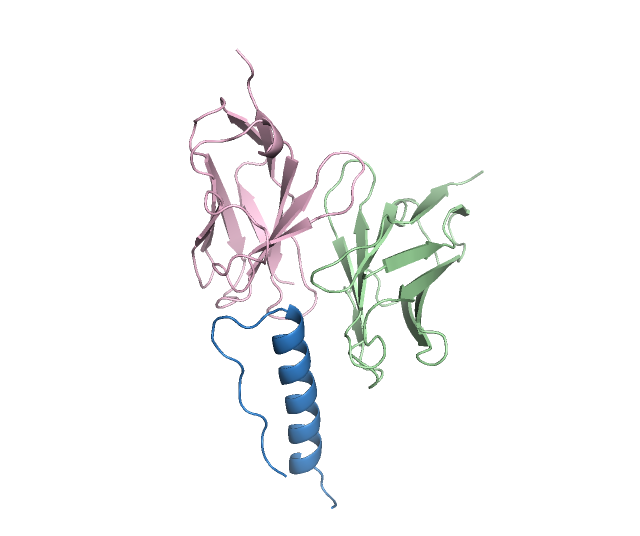
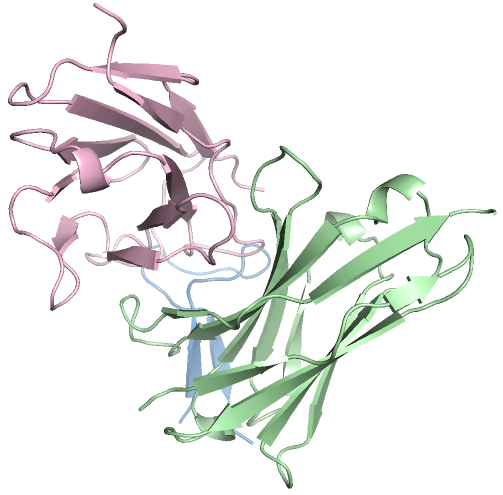
Figure 3: High-affinity antibody designs against antigens, with novelty scores validating PPDiff's capacity for novel design.
Analysis and Scalability
Detailed ablation studies underscore the importance of model components, such as causal attention layers and the number of diffusion steps, which significantly influence design quality. The model's scalability is demonstrated through an increase in performance with larger architectures, suggesting future enhancements could focus on scaling model parameters and datasets further.
Moreover, exploring informative priors, like additional pretraining datasets, illustrated that while Swiss-Prot data did not significantly enhance performance, the approach sets a foundation for integrating diverse data sources in future iterations.
Conclusion
PPDiff stands out as an effective model for designing protein-protein interactions by addressing prior limitations in sequence and structural design. Its robust architecture and superior performance metrics suggest promising directions for future developments in protein engineering, particularly in therapeutic design, where high-affinity binders are crucial. Subsequent research could validate these findings through wet-lab experiments to establish its utility in real-world biomedical applications.